

Drug resistance in Plasmodium
- PMID: 29355852
- PMCID: PMC6371404
- DOI: 10.1038/nrmicro.2017.161
Drug resistance in Plasmodium
Abstract
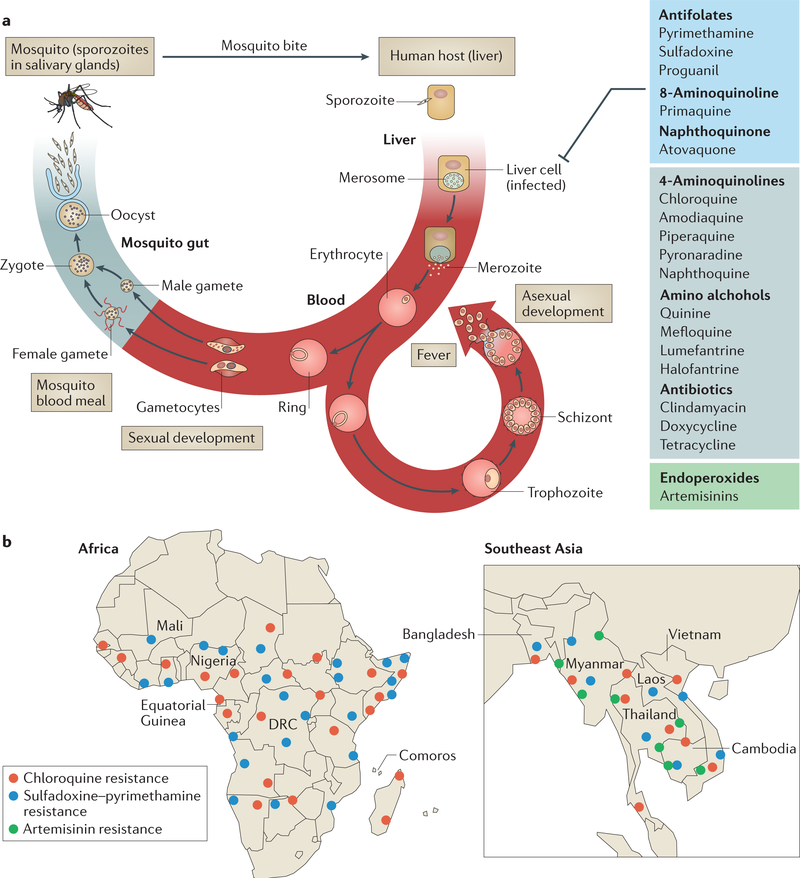
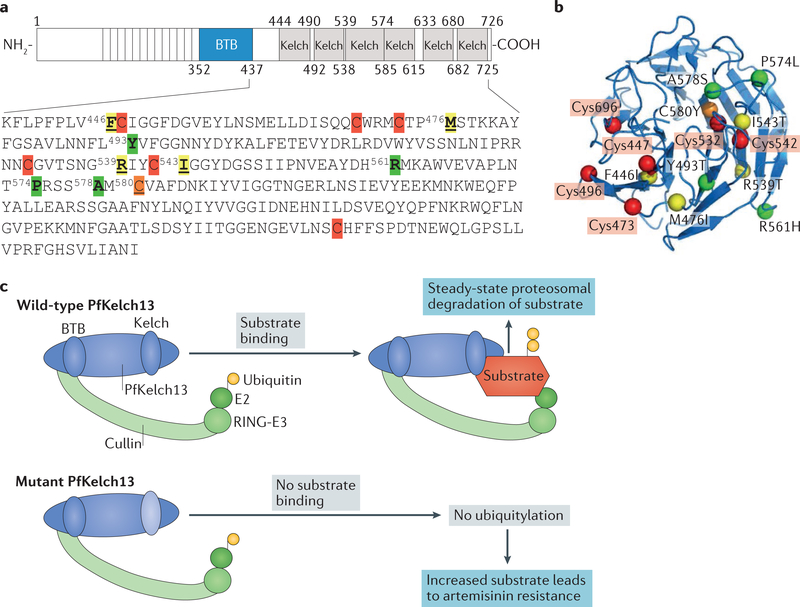
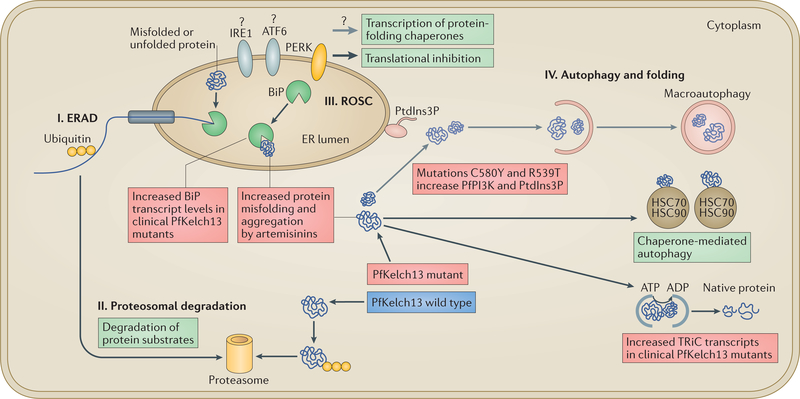
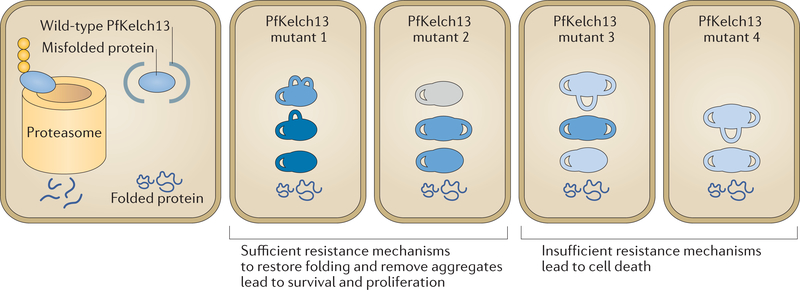
A marked decrease in malaria-related deaths worldwide has been attributed to the administration of effective antimalarials against Plasmodium falciparum, in particular, artemisinin-based combination therapies (ACTs). Increasingly, ACTs are also used to treat Plasmodium vivax, the second major human malaria parasite. However, resistance to frontline artemisinins and partner drugs is now causing the failure of P. falciparum ACTs in southeast Asia. In this Review, we discuss our current knowledge of markers and mechanisms of resistance to artemisinins and ACTs. In particular, we describe the identification of mutations in the propeller domains of Kelch 13 as the primary marker for artemisinin resistance in P. falciparum and explore two major mechanisms of resistance that have been independently proposed: the activation of the unfolded protein response and proteostatic dysregulation of parasite phosphatidylinositol 3- kinase. We emphasize the continuing challenges and the imminent need to understand mechanisms of resistance to improve parasite detection strategies, develop new combinations to eliminate resistant parasites and prevent their global spread.
Figures
References
-
- World Health Organization. World Malaria Report 2015 (WHO, 2015).
-
- World Health Organization. World Malaria Report 2016 (WHO, 2016).
-
- Haldar K, Murphy SC, Milner DA & Taylor TE Malaria: mechanisms of erythrocytic infection and pathological correlates of severe disease. Annu. Rev. Pathol 2, 217–249 (2007). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
