

Clinical, Biomarker, and Molecular Delineations and Genotype-Phenotype Correlations of Ataxia With Oculomotor Apraxia Type 1
- PMID: 29356829
- PMCID: PMC5933354
- DOI: 10.1001/jamaneurol.2017.4373
Clinical, Biomarker, and Molecular Delineations and Genotype-Phenotype Correlations of Ataxia With Oculomotor Apraxia Type 1
Abstract
Importance: Ataxia with oculomotor apraxia type 1 (AOA1) is an autosomal recessive cerebellar ataxia due to mutations in the aprataxin gene (APTX) that is characterized by early-onset cerebellar ataxia, oculomotor apraxia, axonal motor neuropathy, and eventual decrease of albumin serum levels.
Objectives: To improve the clinical, biomarker, and molecular delineation of AOA1 and provide genotype-phenotype correlations.
Design, setting, and participants: This retrospective analysis included the clinical, biological (especially regarding biomarkers of the disease), electrophysiologic, imaging, and molecular data of all patients consecutively diagnosed with AOA1 in a single genetics laboratory from January 1, 2002, through December 31, 2014. Data were analyzed from January 1, 2015, through January 31, 2016.
Main outcomes and measures: The clinical, biological, and molecular spectrum of AOA1 and genotype-phenotype correlations.
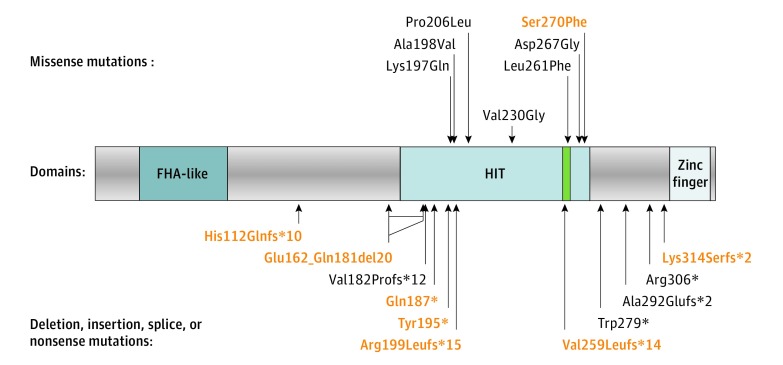
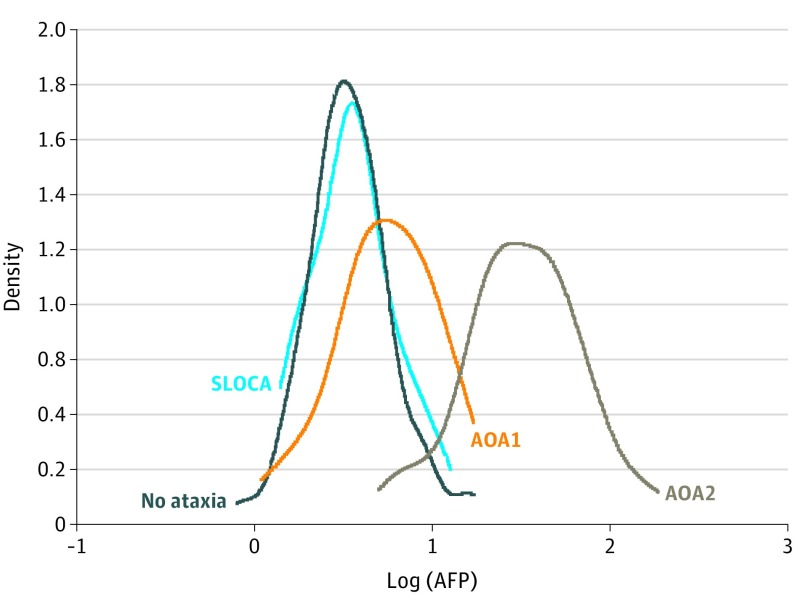
Results: The diagnosis of AOA1 was confirmed in 80 patients (46 men [58%] and 34 women [42%]; mean [SD] age at onset, 7.7 [7.4] years) from 51 families, including 57 new (with 8 new mutations) and 23 previously described patients. Elevated levels of α-fetoprotein (AFP) were found in 33 patients (41%); hypoalbuminemia, in 50 (63%). Median AFP level was higher in patients with AOA1 (6.0 ng/mL; range, 1.1-17.0 ng/mL) than in patients without ataxia (3.4 ng/mL; range, 0.8-17.2 ng/mL; P < .01). Decreased albumin levels (ρ = -0.532) and elevated AFP levels (ρ = 0.637) were correlated with disease duration. The p.Trp279* mutation, initially reported as restricted to the Portuguese founder haplotype, was discovered in 53 patients with AOA1 (66%) with broad white racial origins. Oculomotor apraxia was found in 49 patients (61%); polyneuropathy, in 74 (93%); and cerebellar atrophy, in 78 (98%). Oculomotor apraxia correlated with the severity of ataxia and mutation type, being more frequent with deletion or truncating mutations (83%) than with presence of at least 1 missense variant (17%; P < .01). Mean (SD) age at onset was higher for patients with at least 1 missense mutation (17.7 [11.4] vs 5.2 [2.6] years; P < .001).
Conclusions and relevance: The AFP level, slightly elevated in a substantial fraction of patients, may constitute a new biomarker for AOA1. Oculomotor apraxia may be an optional finding in AOA1 and correlates with more severe disease. The p.Trp279* mutation is the most frequent APTX mutation in the white population. APTX missense mutations may be associated with a milder phenotype.
Conflict of interest statement
Figures
References
-
- Anheim M, Tranchant C, Koenig M. The autosomal recessive cerebellar ataxias. N Engl J Med. 2012;366(7):636-646. - PubMed
-
- Moreira MC, Barbot C, Tachi N, et al. The gene mutated in ataxia-ocular apraxia 1 encodes the new HIT/Zn-finger protein aprataxin. Nat Genet. 2001;29(2):189-193. - PubMed
-
- Date H, Onodera O, Tanaka H, et al. Early-onset ataxia with ocular motor apraxia and hypoalbuminemia is caused by mutations in a new HIT superfamily gene. Nat Genet. 2001;29(2):184-188. - PubMed
-
- Aicardi J, Barbosa C, Andermann E, et al. Ataxia-ocular motor apraxia: a syndrome mimicking ataxia-telangiectasia. Ann Neurol. 1988;24(4):497-502. - PubMed
-
- Le Ber I, Moreira M-C, Rivaud-Péchoux S, et al. Cerebellar ataxia with oculomotor apraxia type 1: clinical and genetic studies. Brain. 2003;126(pt 12):2761-2772. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
