

Triglyceride Metabolism in the Liver
- PMID: 29357123
- PMCID: PMC6376873
- DOI: 10.1002/cphy.c170012
Triglyceride Metabolism in the Liver
Abstract
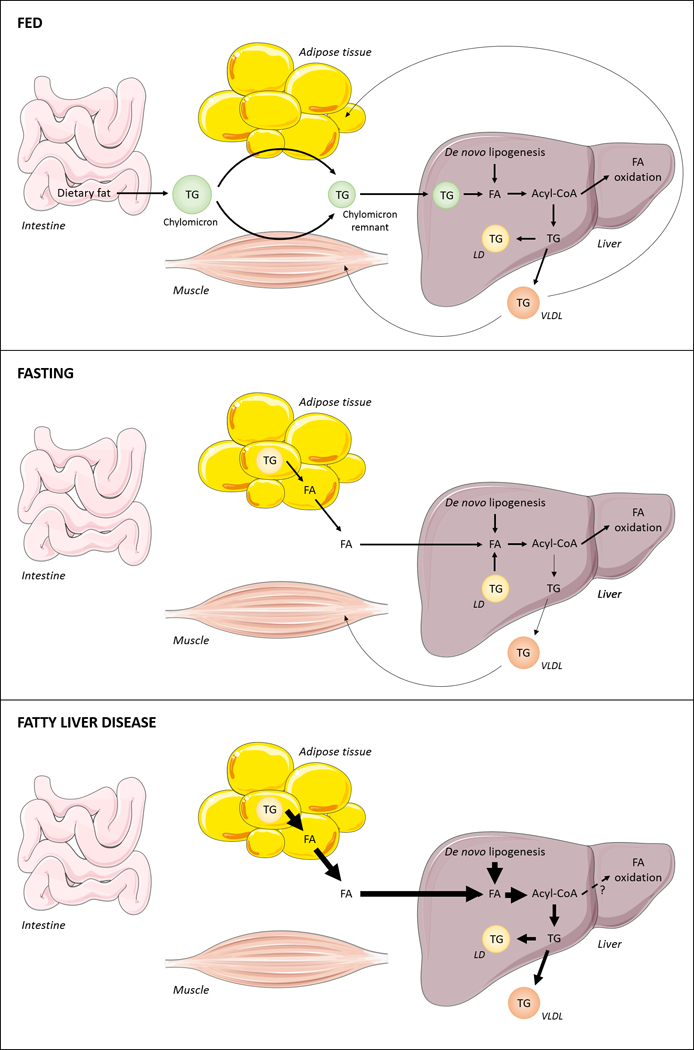
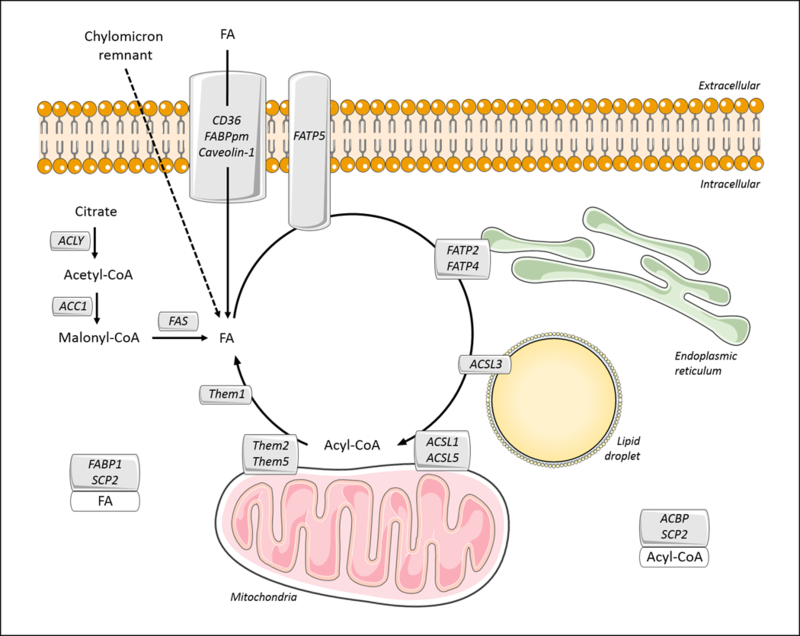
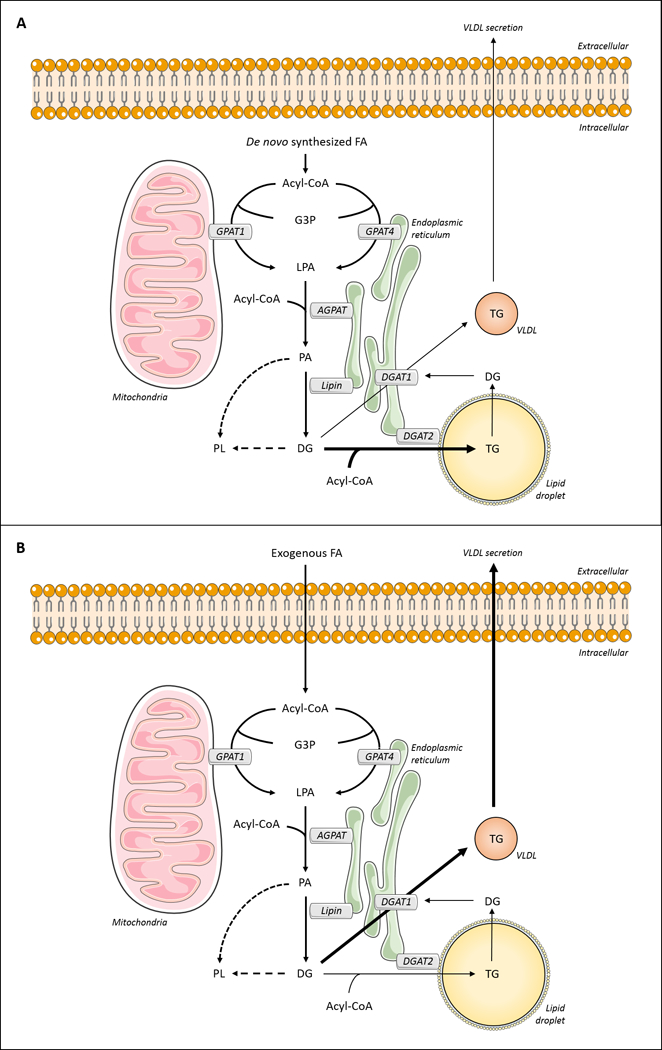
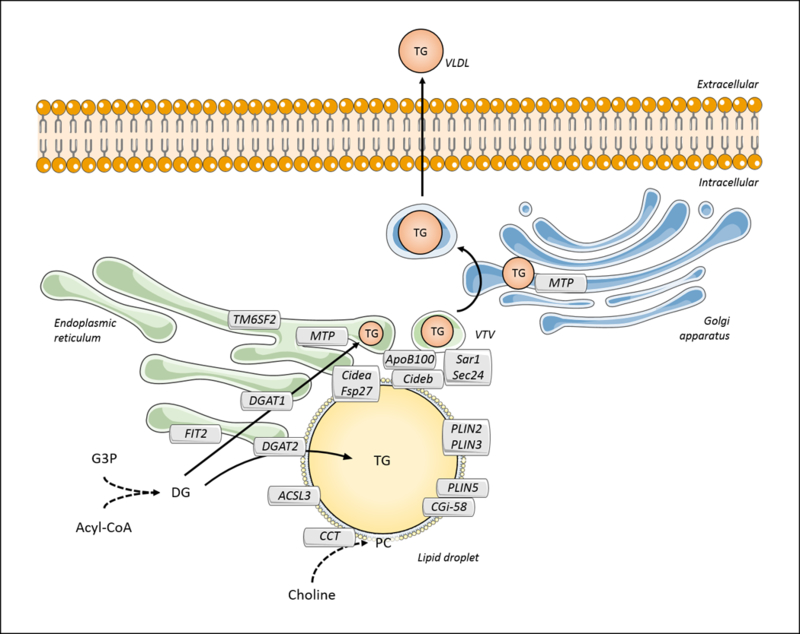
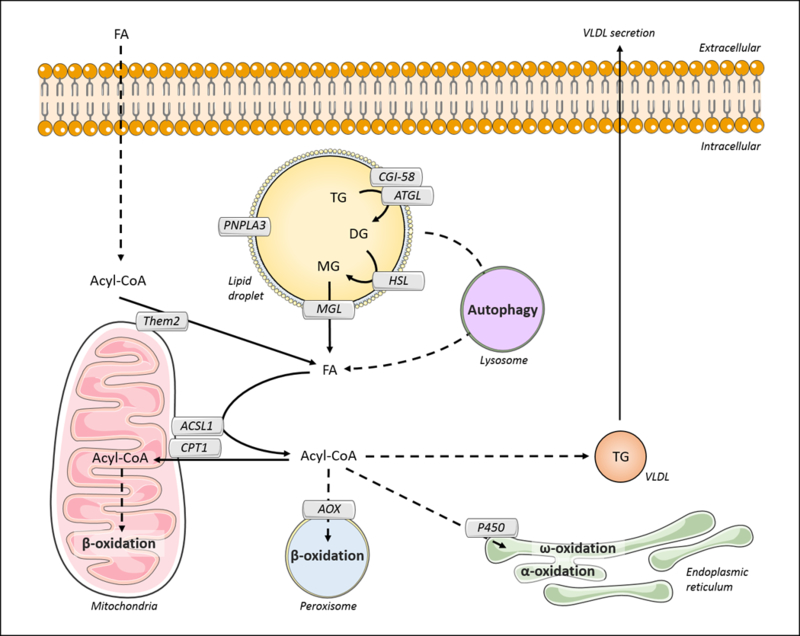
Triglyceride molecules represent the major form of storage and transport of fatty acids within cells and in the plasma. The liver is the central organ for fatty acid metabolism. Fatty acids accrue in liver by hepatocellular uptake from the plasma and by de novo biosynthesis. Fatty acids are eliminated by oxidation within the cell or by secretion into the plasma within triglyceride-rich very low-density lipoproteins. Notwithstanding high fluxes through these pathways, under normal circumstances the liver stores only small amounts of fatty acids as triglycerides. In the setting of overnutrition and obesity, hepatic fatty acid metabolism is altered, commonly leading to the accumulation of triglycerides within hepatocytes, and to a clinical condition known as nonalcoholic fatty liver disease (NAFLD). In this review, we describe the current understanding of fatty acid and triglyceride metabolism in the liver and its regulation in health and disease, identifying potential directions for future research. Advances in understanding the molecular mechanisms underlying the hepatic fat accumulation are critical to the development of targeted therapies for NAFLD. © 2018 American Physiological Society. Compr Physiol 8:1-22, 2018.
Copyright © 2018 John Wiley & Sons, Inc.
Figures
References
-
- Abo-Hashema KAH, Cake MH, Power GW, and Clarke D. Evidence for triacylglycerol synthesis in the lumen of microsomes via a lipolysis-esterification pathway involving carnitine acyltransferases. J Biol Chem 274: 35577–35582, 1999. - PubMed
-
- Abu-Elheiga L, Matzuk MM, Abo-Hashema KA, and Wakil SJ. Continuous fatty acid oxidation and reduced fat storage in mice lacking acetyl-CoA carboxylase 2. Science 291: 2613–2616, 2001. - PubMed
-
- Achouri Y, Hegarty BD, Allanic D, Becard D, Hainault I, Ferre P, and Foufelle F. Long chain fatty acyl-CoA synthetase 5 expression is induced by insulin and glucose: Involvement of sterol regulatory element-binding protein-1c. Biochimie 87: 1149–1155, 2005. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
