

Telomerecat: A ploidy-agnostic method for estimating telomere length from whole genome sequencing data
- PMID: 29358629
- PMCID: PMC5778012
- DOI: 10.1038/s41598-017-14403-y
Telomerecat: A ploidy-agnostic method for estimating telomere length from whole genome sequencing data
Erratum in
-
Publisher Correction: Telomerecat: A ploidy-agnostic method for estimating telomere length from whole genome sequencing data.Sci Rep. 2018 Sep 3;8(1):13376. doi: 10.1038/s41598-018-31524-0. Sci Rep. 2018. PMID: 30177810 Free PMC article.
Abstract
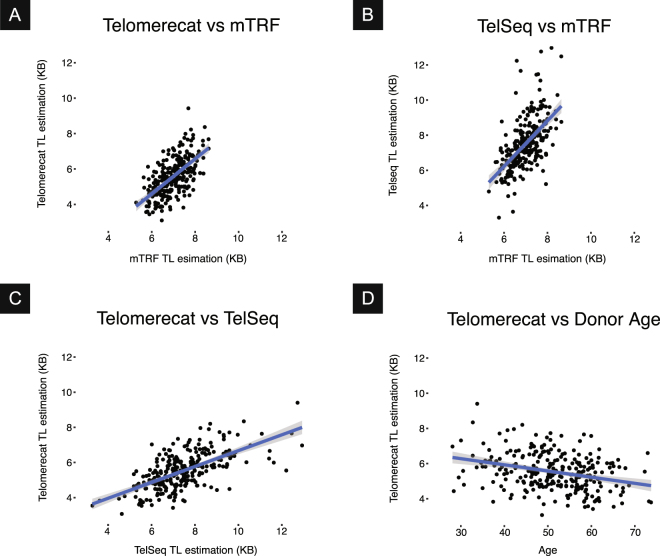
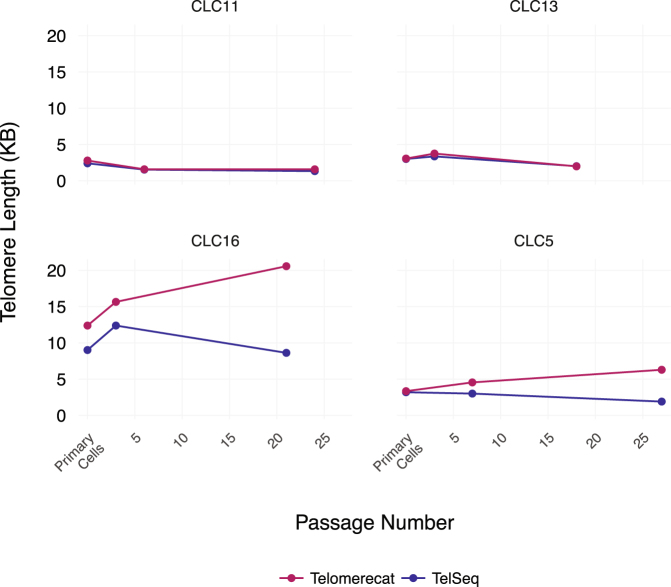
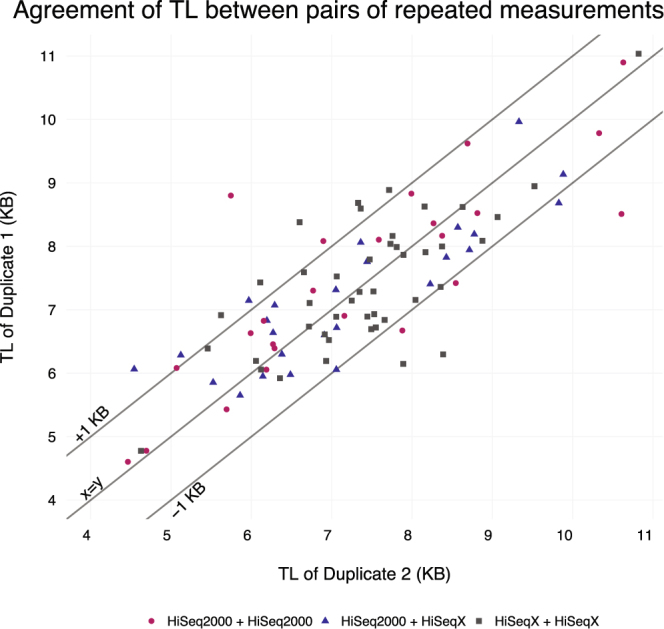
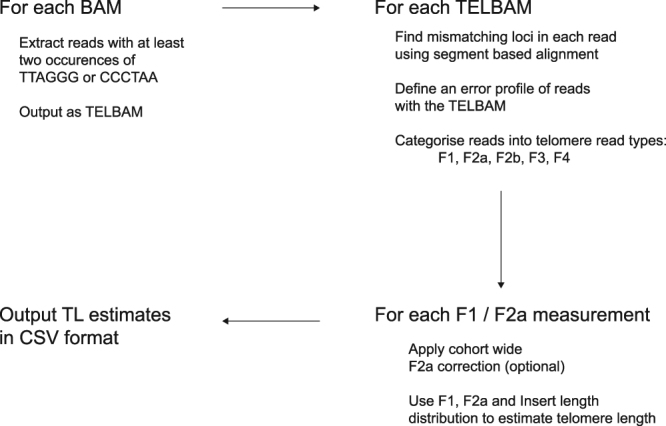
Telomere length is a risk factor in disease and the dynamics of telomere length are crucial to our understanding of cell replication and vitality. The proliferation of whole genome sequencing represents an unprecedented opportunity to glean new insights into telomere biology on a previously unimaginable scale. To this end, a number of approaches for estimating telomere length from whole-genome sequencing data have been proposed. Here we present Telomerecat, a novel approach to the estimation of telomere length. Previous methods have been dependent on the number of telomeres present in a cell being known, which may be problematic when analysing aneuploid cancer data and non-human samples. Telomerecat is designed to be agnostic to the number of telomeres present, making it suited for the purpose of estimating telomere length in cancer studies. Telomerecat also accounts for interstitial telomeric reads and presents a novel approach to dealing with sequencing errors. We show that Telomerecat performs well at telomere length estimation when compared to leading experimental and computational methods. Furthermore, we show that it detects expected patterns in longitudinal data, repeated measurements, and cross-species comparisons. We also apply the method to a cancer cell data, uncovering an interesting relationship with the underlying telomerase genotype.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures



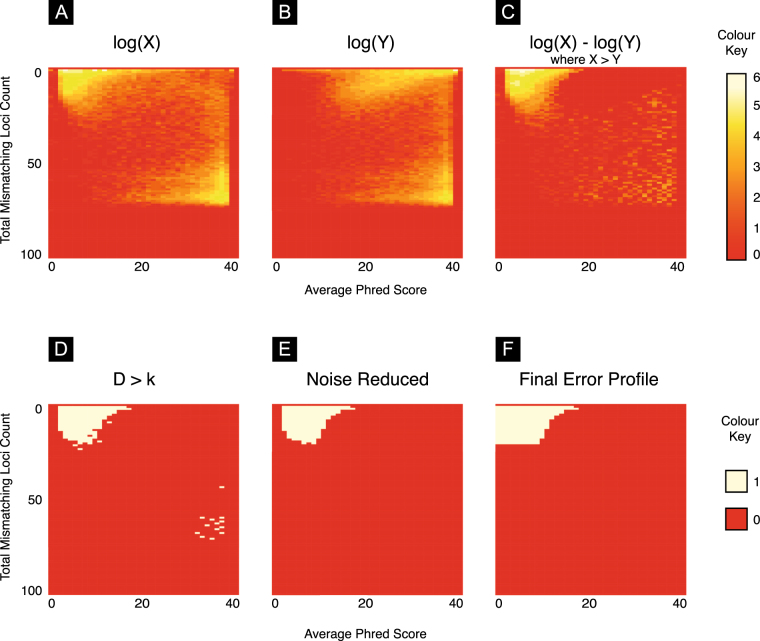
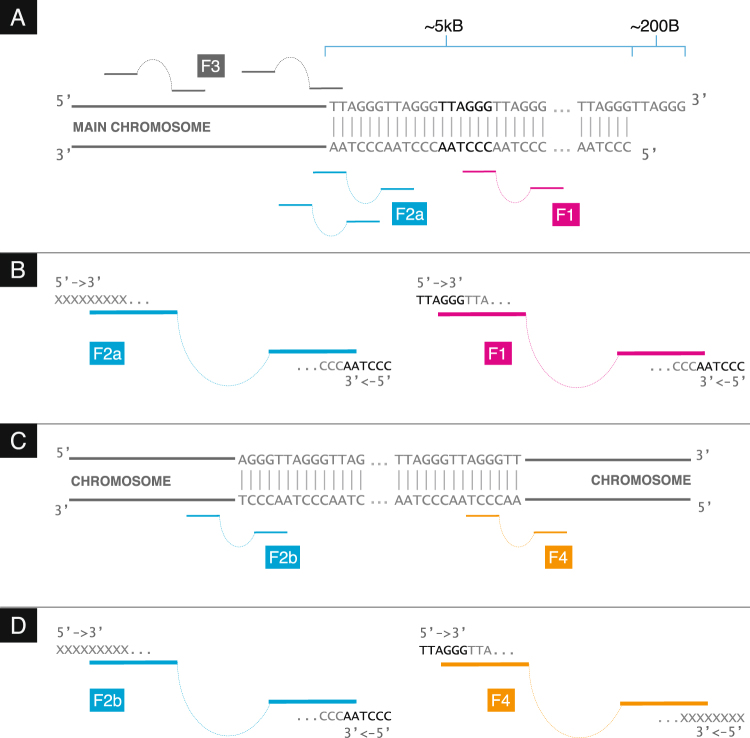

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
