

Concussion, microvascular injury, and early tauopathy in young athletes after impact head injury and an impact concussion mouse model
- PMID: 29360998
- PMCID: PMC5837414
- DOI: 10.1093/brain/awx350
Concussion, microvascular injury, and early tauopathy in young athletes after impact head injury and an impact concussion mouse model
Abstract
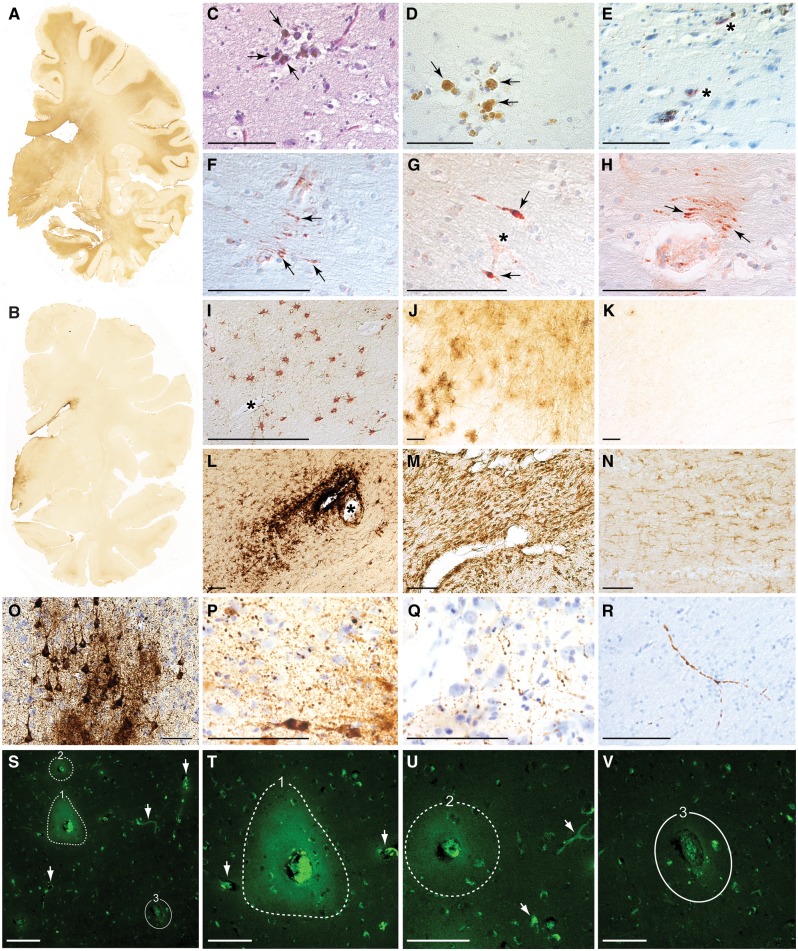
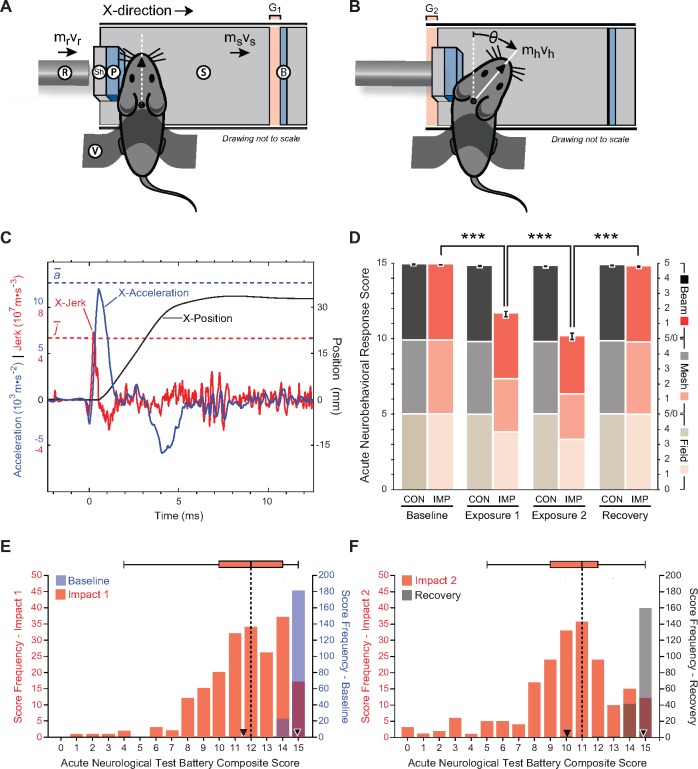
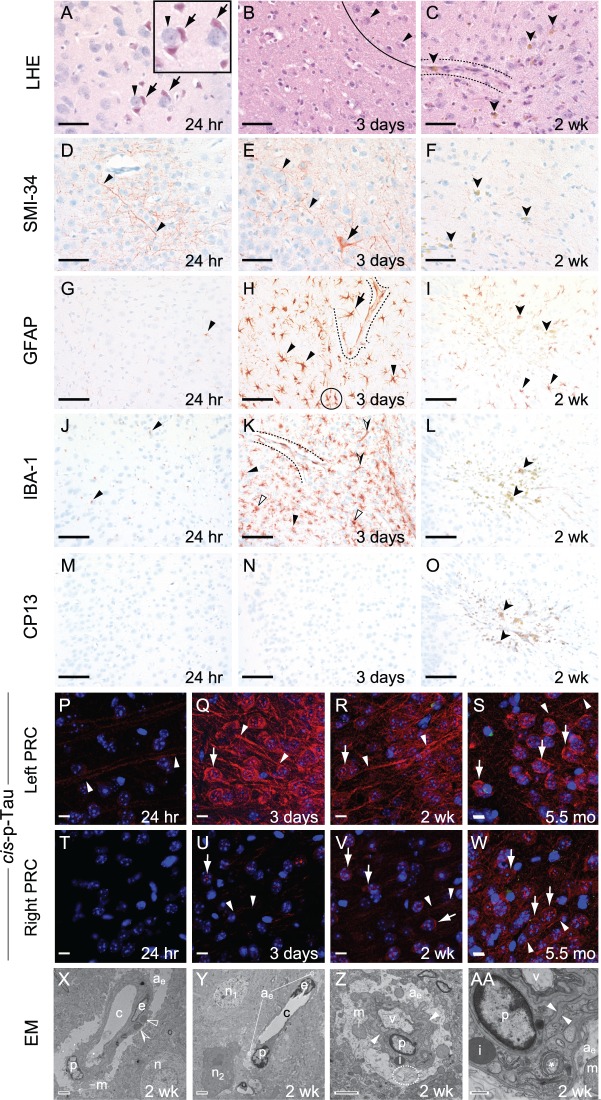
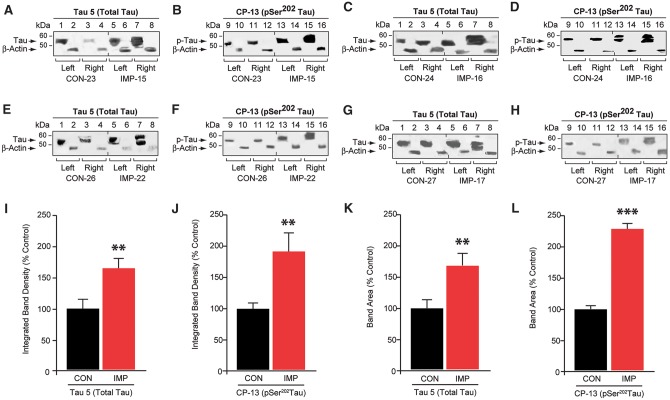
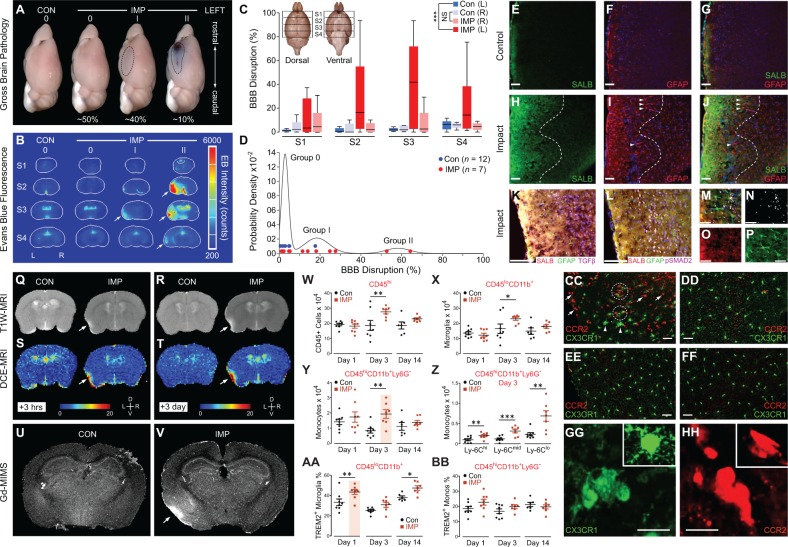
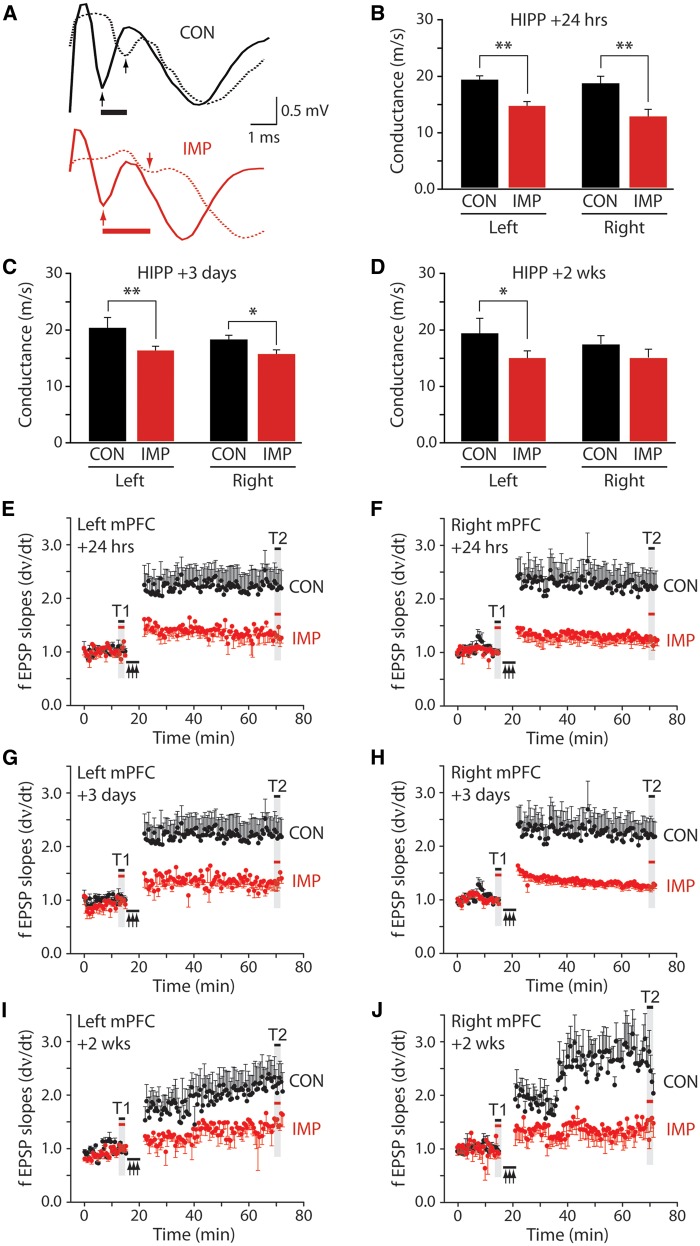
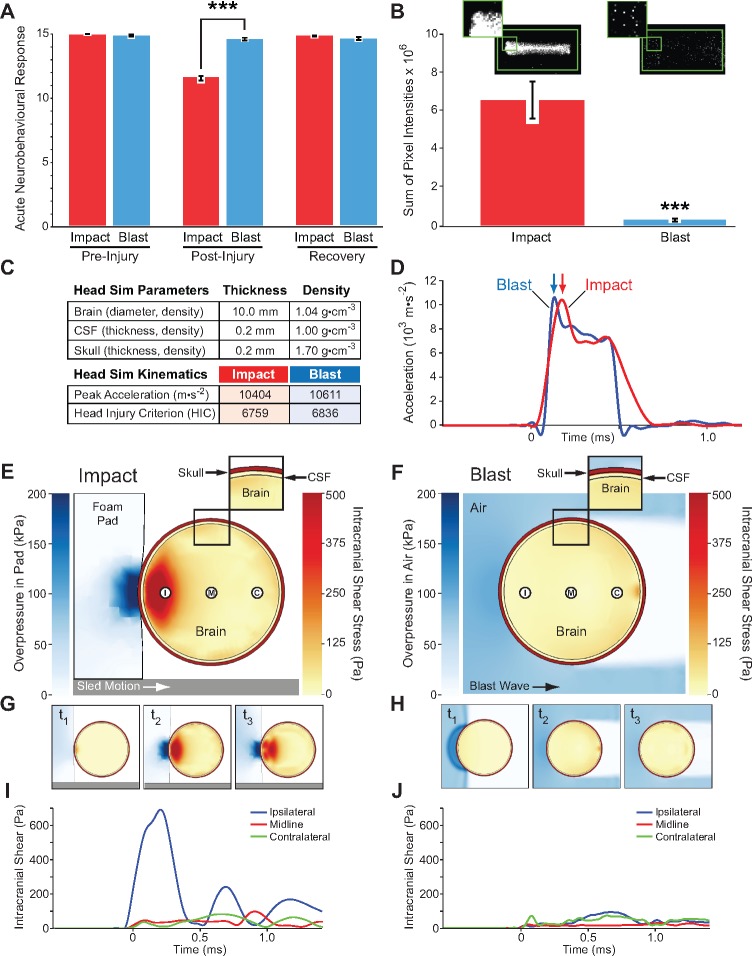
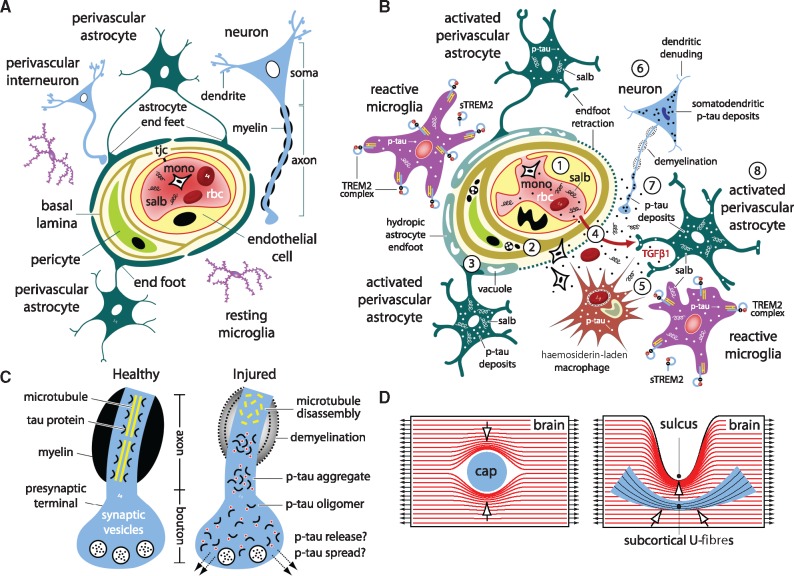
The mechanisms underpinning concussion, traumatic brain injury, and chronic traumatic encephalopathy, and the relationships between these disorders, are poorly understood. We examined post-mortem brains from teenage athletes in the acute-subacute period after mild closed-head impact injury and found astrocytosis, myelinated axonopathy, microvascular injury, perivascular neuroinflammation, and phosphorylated tau protein pathology. To investigate causal mechanisms, we developed a mouse model of lateral closed-head impact injury that uses momentum transfer to induce traumatic head acceleration. Unanaesthetized mice subjected to unilateral impact exhibited abrupt onset, transient course, and rapid resolution of a concussion-like syndrome characterized by altered arousal, contralateral hemiparesis, truncal ataxia, locomotor and balance impairments, and neurobehavioural deficits. Experimental impact injury was associated with axonopathy, blood-brain barrier disruption, astrocytosis, microgliosis (with activation of triggering receptor expressed on myeloid cells, TREM2), monocyte infiltration, and phosphorylated tauopathy in cerebral cortex ipsilateral and subjacent to impact. Phosphorylated tauopathy was detected in ipsilateral axons by 24 h, bilateral axons and soma by 2 weeks, and distant cortex bilaterally at 5.5 months post-injury. Impact pathologies co-localized with serum albumin extravasation in the brain that was diagnostically detectable in living mice by dynamic contrast-enhanced MRI. These pathologies were also accompanied by early, persistent, and bilateral impairment in axonal conduction velocity in the hippocampus and defective long-term potentiation of synaptic neurotransmission in the medial prefrontal cortex, brain regions distant from acute brain injury. Surprisingly, acute neurobehavioural deficits at the time of injury did not correlate with blood-brain barrier disruption, microgliosis, neuroinflammation, phosphorylated tauopathy, or electrophysiological dysfunction. Furthermore, concussion-like deficits were observed after impact injury, but not after blast exposure under experimental conditions matched for head kinematics. Computational modelling showed that impact injury generated focal point loading on the head and seven-fold greater peak shear stress in the brain compared to blast exposure. Moreover, intracerebral shear stress peaked before onset of gross head motion. By comparison, blast induced distributed force loading on the head and diffuse, lower magnitude shear stress in the brain. We conclude that force loading mechanics at the time of injury shape acute neurobehavioural responses, structural brain damage, and neuropathological sequelae triggered by neurotrauma. These results indicate that closed-head impact injuries, independent of concussive signs, can induce traumatic brain injury as well as early pathologies and functional sequelae associated with chronic traumatic encephalopathy. These results also shed light on the origins of concussion and relationship to traumatic brain injury and its aftermath.awx350media15713427811001.
Keywords: TREM2; chronic traumatic encephalopathy; concussion; tau protein; traumatic brain injury.
© The Author(s) (2018). Published by Oxford University Press on behalf of the Guarantors of Brain.
Figures
References
-
- Abbas K, Shenk TE, Poole VN, Breedlove EL, Leverenz LJ, Nauman EA, et al. Alteration of default mode network in high school football athletes due to repetitive subconcussive mild traumatic brain injury: a resting-state functional magnetic resonance imaging study. Brain Connect 2015; 5: 91–101. - PubMed
-
- Adams JH, Graham DI, Murray LS, Scott G. Diffuse axonal injury due to nonmissile head injury in humans: an analysis of 45 cases. Ann Neurol 1982; 12: 557–63. - PubMed
-
- Annegers JF, Hauser WA, Coan SP, Rocca WA. A population-based study of seizures after traumatic brain injuries. N Eng J Med 1998; 338: 20–4. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- F31 NS080564/NS/NINDS NIH HHS/United States
- S10 RR024523/RR/NCRR NIH HHS/United States
- F31 NS077796/NS/NINDS NIH HHS/United States
- R01 AG029385/AG/NIA NIH HHS/United States
- U01 NS086659/NS/NINDS NIH HHS/United States
- P30 AG013846/AG/NIA NIH HHS/United States
- U01 NS096835/NS/NINDS NIH HHS/United States
- CIHR/Canada
- T32 EB006359/EB/NIBIB NIH HHS/United States
- I01 CX001038/CX/CSRD VA/United States
- R01 NS044421/NS/NINDS NIH HHS/United States
- I01 RX002170/RX/RRD VA/United States
- R01 NS032151/NS/NINDS NIH HHS/United States
- R01 AG055559/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
