

Estimating Time to the Common Ancestor for a Beneficial Allele
- PMID: 29361025
- PMCID: PMC5888984
- DOI: 10.1093/molbev/msy006
Estimating Time to the Common Ancestor for a Beneficial Allele
Erratum in
-
Molecular Biology and Evolution, Volume 35, Issue 4.Mol Biol Evol. 2018 Jul 1;35(7):1821. doi: 10.1093/molbev/msy057. Mol Biol Evol. 2018. PMID: 29659984 Free PMC article. No abstract available.
Abstract
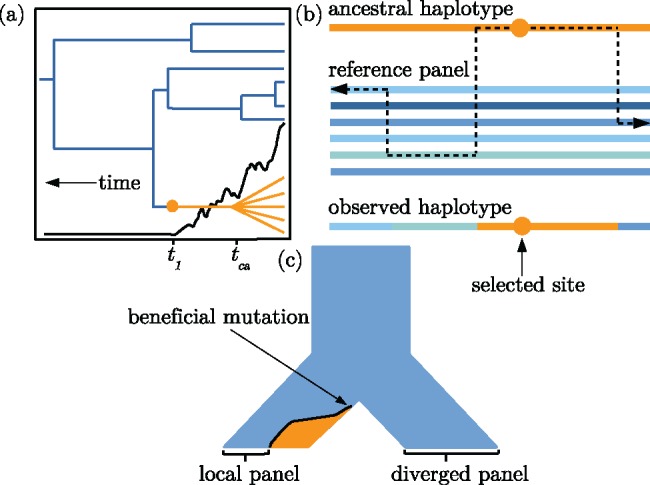
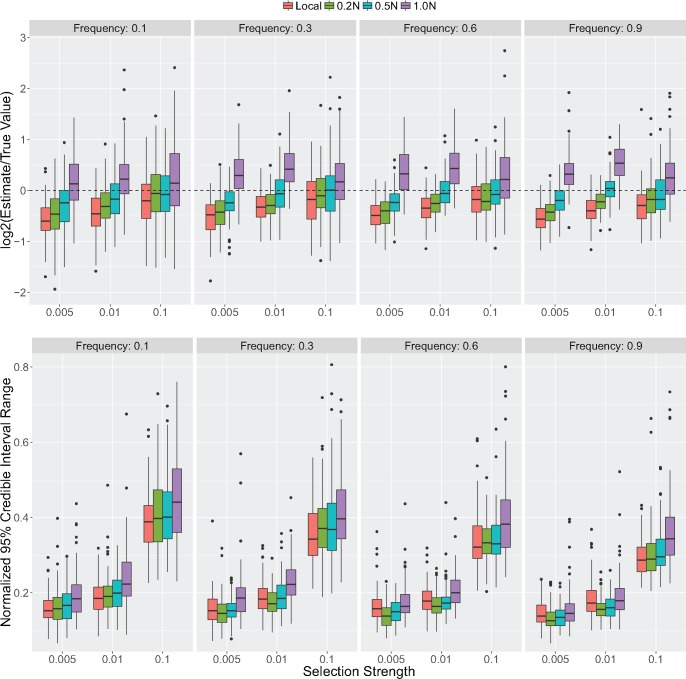
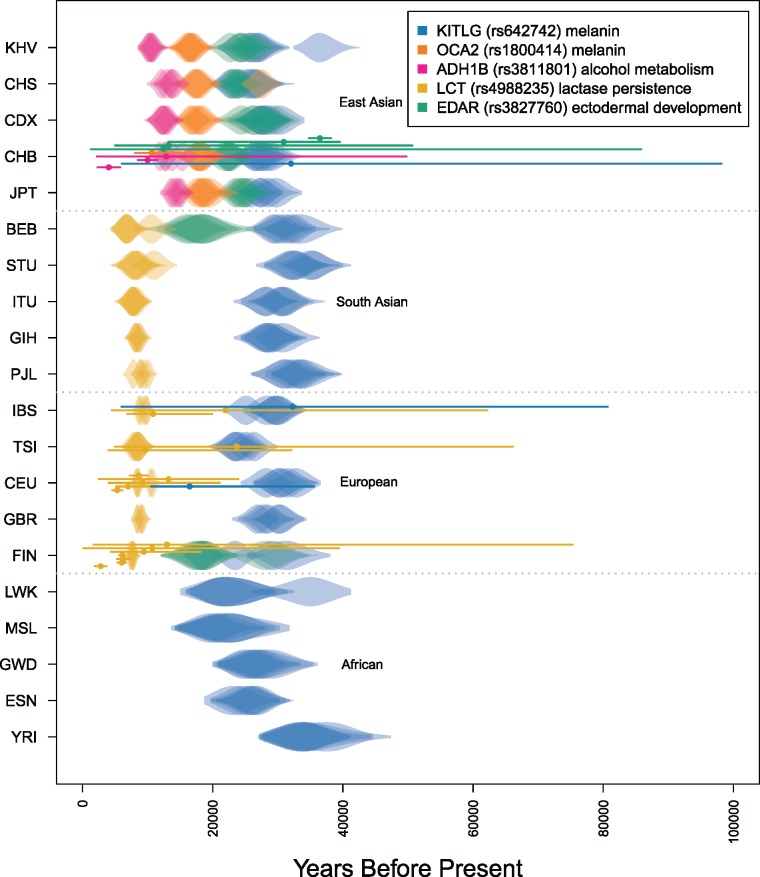
The haplotypes of a beneficial allele carry information about its history that can shed light on its age and the putative cause for its increase in frequency. Specifically, the signature of an allele's age is contained in the pattern of variation that mutation and recombination impose on its haplotypic background. We provide a method to exploit this pattern and infer the time to the common ancestor of a positively selected allele following a rapid increase in frequency. We do so using a hidden Markov model which leverages the length distribution of the shared ancestral haplotype, the accumulation of derived mutations on the ancestral background, and the surrounding background haplotype diversity. Using simulations, we demonstrate how the inclusion of information from both mutation and recombination events increases accuracy relative to approaches that only consider a single type of event. We also show the behavior of the estimator in cases where data do not conform to model assumptions, and provide some diagnostics for assessing and improving inference. Using the method, we analyze population-specific patterns in the 1000 Genomes Project data to estimate the timing of adaptation for several variants which show evidence of recent selection and functional relevance to diet, skin pigmentation, and morphology in humans.
Figures
References
-
- Allentoft ME, Sikora M, Sjögren K-G, Rasmussen S, Rasmussen M, Stenderup J, Damgaard PB, Schroeder H, Ahlström T, Vinner L.. 2015. Population genomics of Bronze Age Eurasia. Nature 5227555: 167–172. - PubMed
-
- Auton A, McVean G.. 2012. Estimating recombination rates from genetic variation in humans In: Anisimova M. ed, Evolutionary genomics. Methods in Molecular Biology (Methods and Protocols), vol 856. New York: Humana Press. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
