

Lack of beta-arrestin signaling in the absence of active G proteins
- PMID: 29362459
- PMCID: PMC5780443
- DOI: 10.1038/s41467-017-02661-3
Lack of beta-arrestin signaling in the absence of active G proteins
Abstract
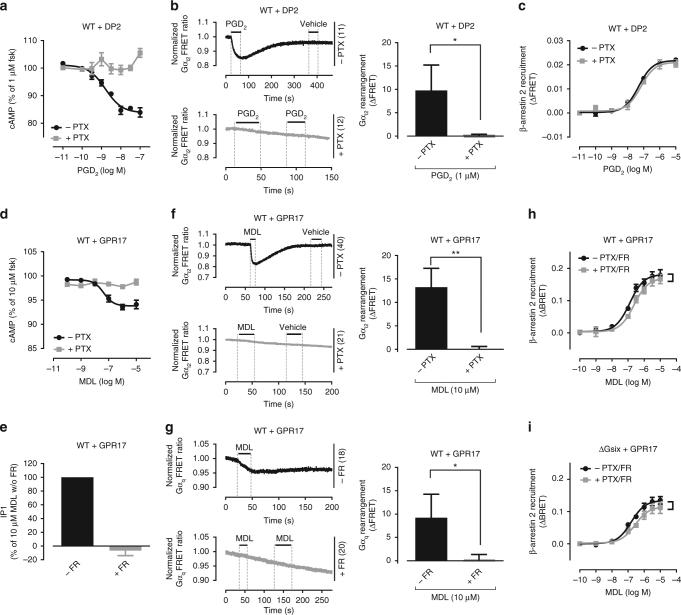
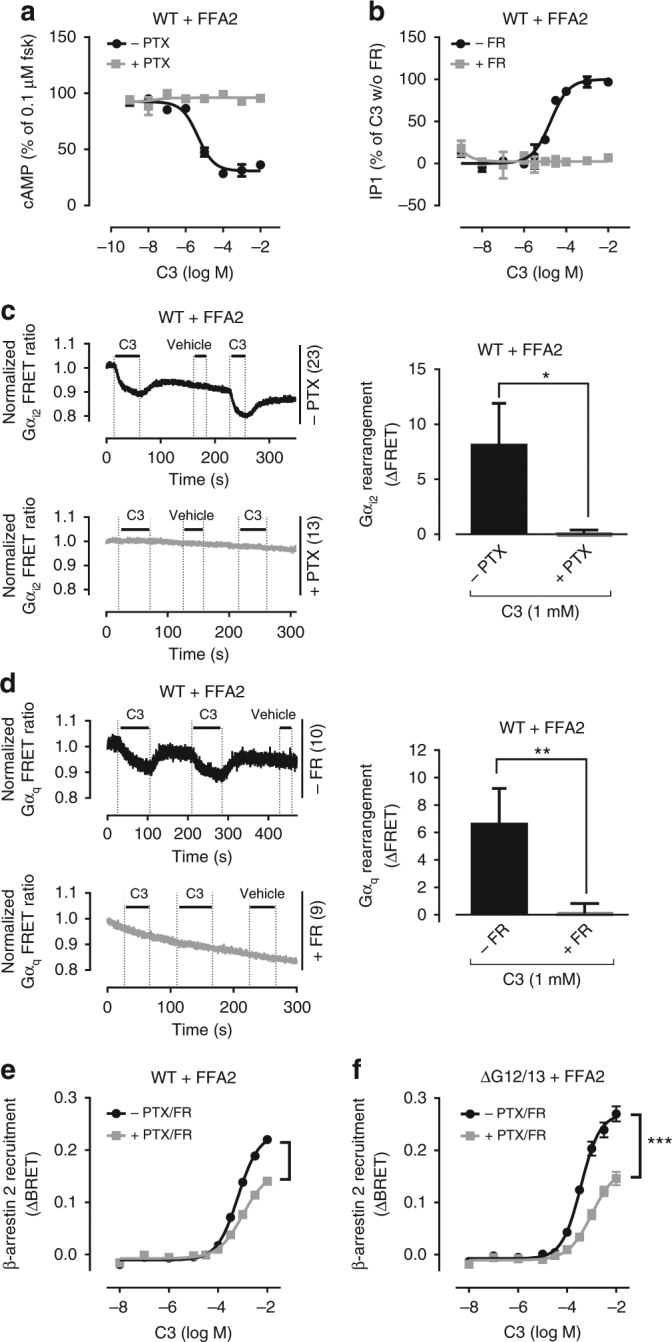
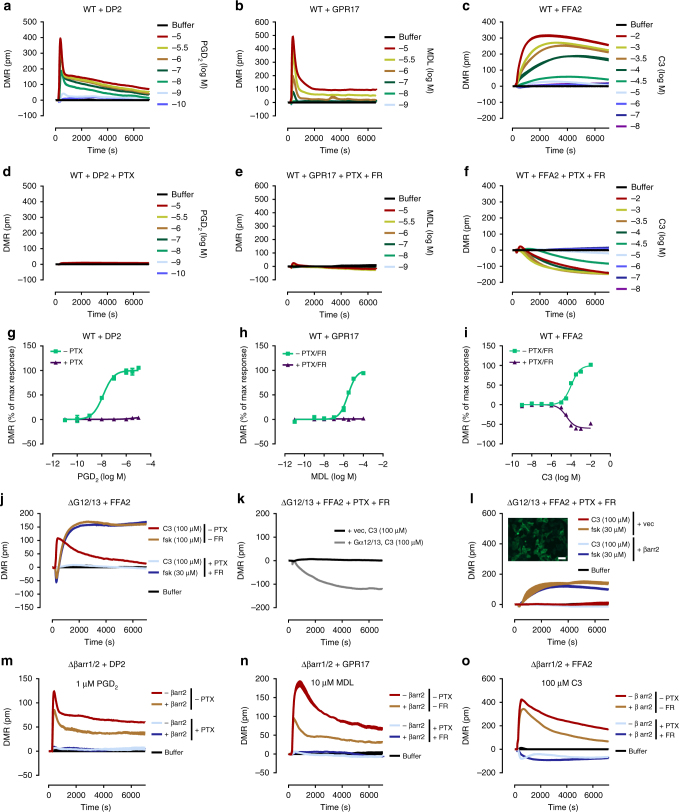
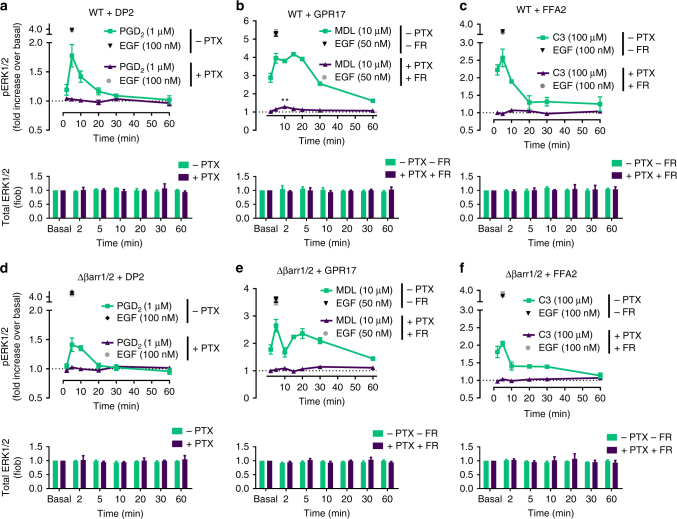
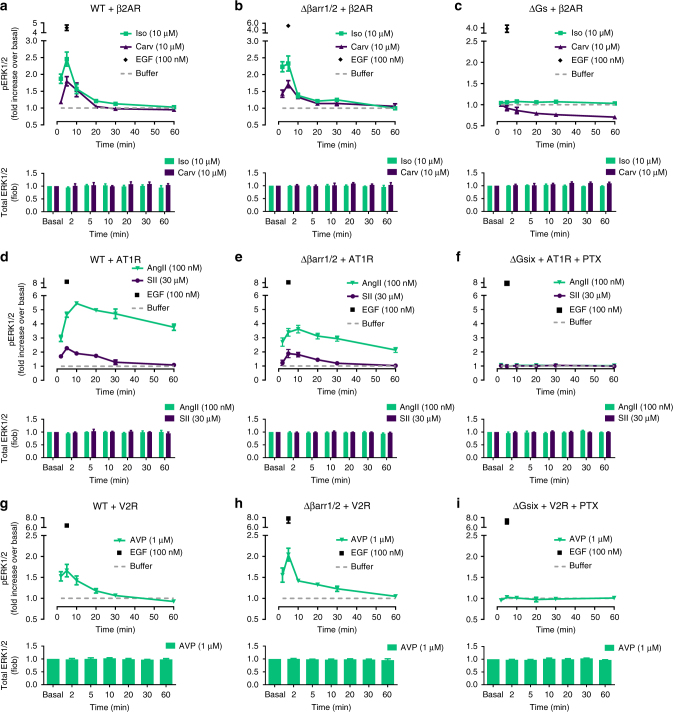
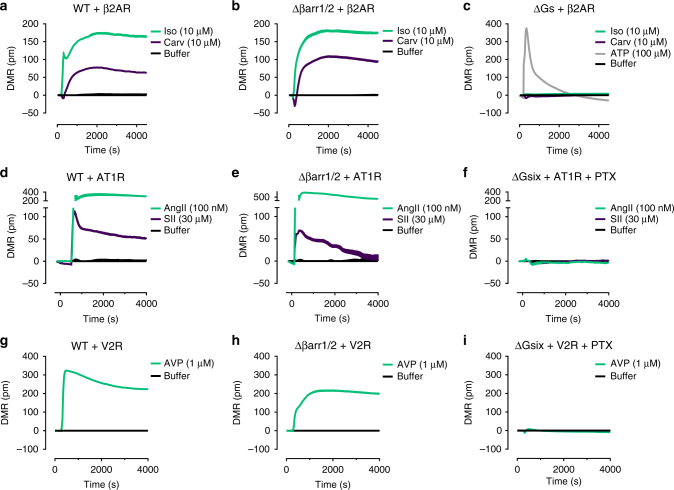
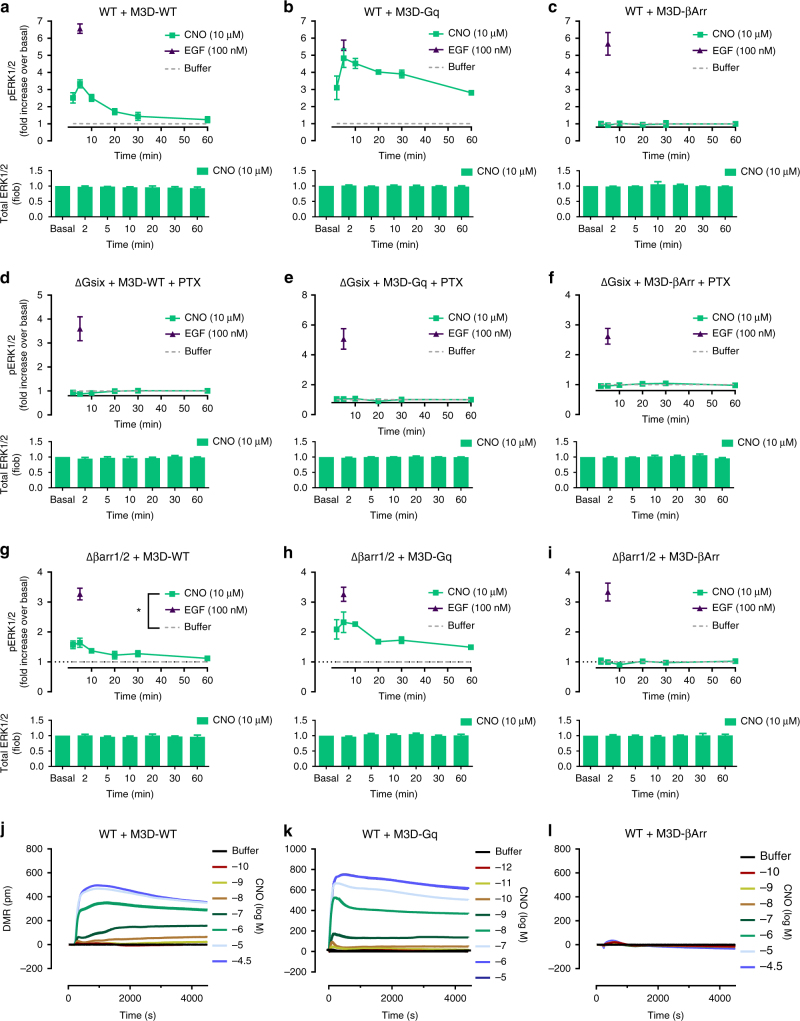
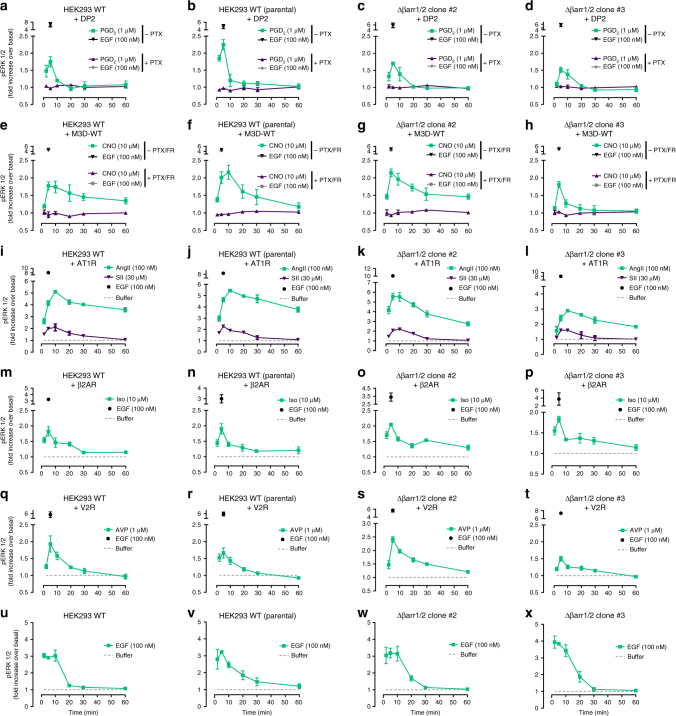
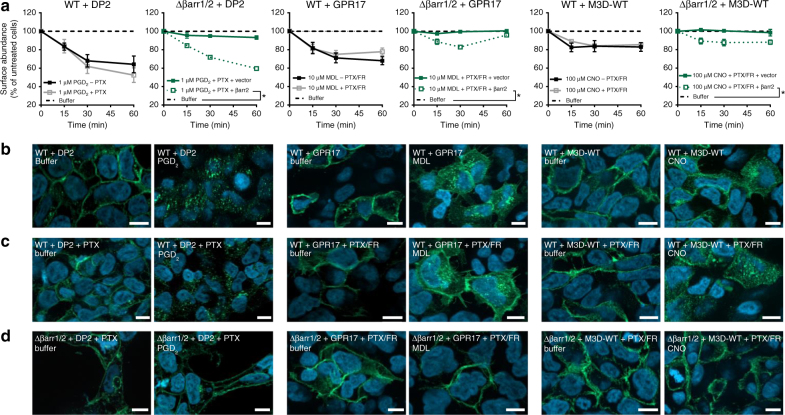
G protein-independent, arrestin-dependent signaling is a paradigm that broadens the signaling scope of G protein-coupled receptors (GPCRs) beyond G proteins for numerous biological processes. However, arrestin signaling in the collective absence of functional G proteins has never been demonstrated. Here we achieve a state of "zero functional G" at the cellular level using HEK293 cells depleted by CRISPR/Cas9 technology of the Gs/q/12 families of Gα proteins, along with pertussis toxin-mediated inactivation of Gi/o. Together with HEK293 cells lacking β-arrestins ("zero arrestin"), we systematically dissect G protein- from arrestin-driven signaling outcomes for a broad set of GPCRs. We use biochemical, biophysical, label-free whole-cell biosensing and ERK phosphorylation to identify four salient features for all receptors at "zero functional G": arrestin recruitment and internalization, but-unexpectedly-complete failure to activate ERK and whole-cell responses. These findings change our understanding of how GPCRs function and in particular of how they activate ERK1/2.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Benovic JL, et al. Functional desensitization of the isolated beta-adrenergic receptor by the beta-adrenergic receptor kinase: potential role of an analog of the retinal protein arrestin (48-kDa protein) Proc. Natl Acad. Sci. USA. 1987;84:8879–8882. doi: 10.1073/pnas.84.24.8879. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
