

Biased signaling by thyroid-stimulating hormone receptor-specific antibodies determines thyrocyte survival in autoimmunity
- PMID: 29363585
- PMCID: PMC5812361
- DOI: 10.1126/scisignal.aah4120
Biased signaling by thyroid-stimulating hormone receptor-specific antibodies determines thyrocyte survival in autoimmunity
Abstract
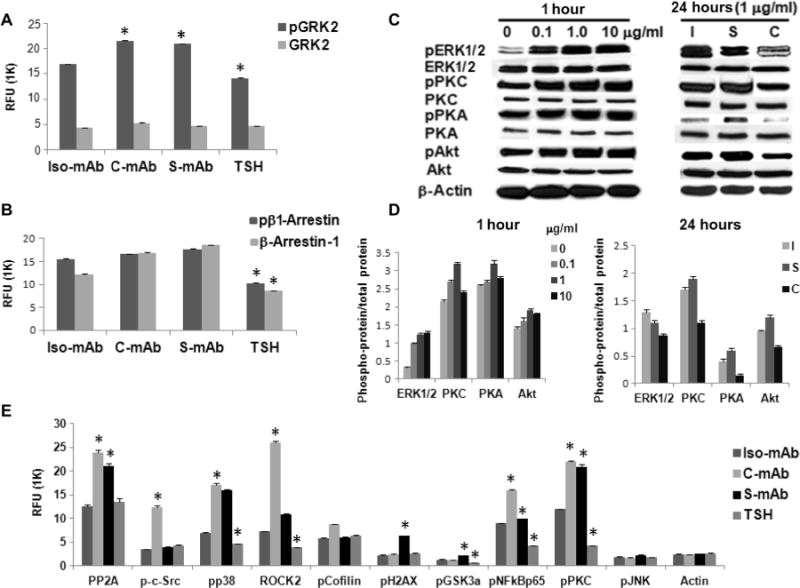
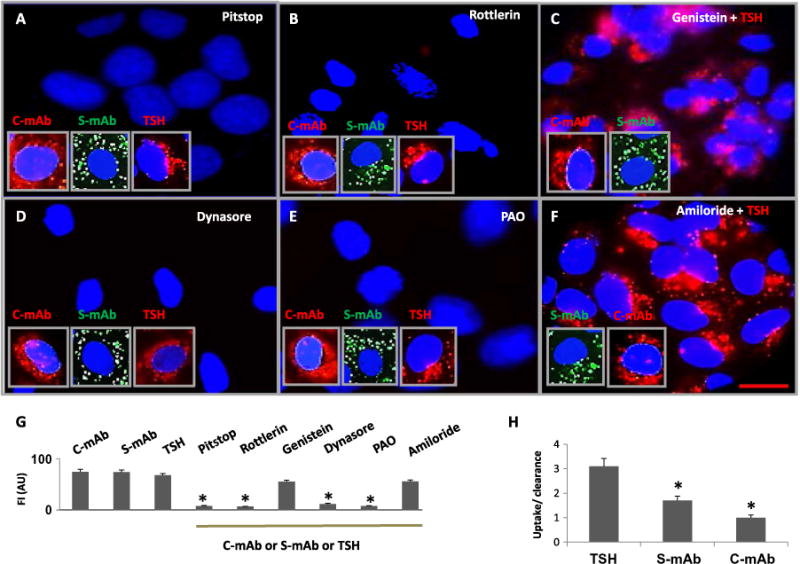
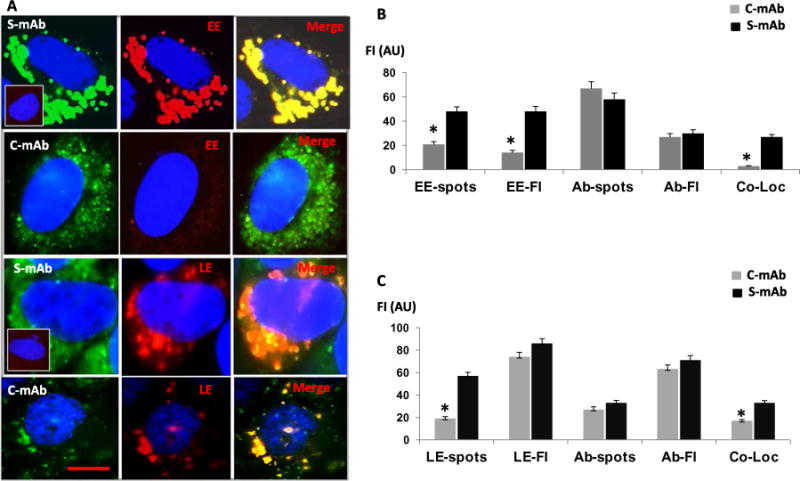
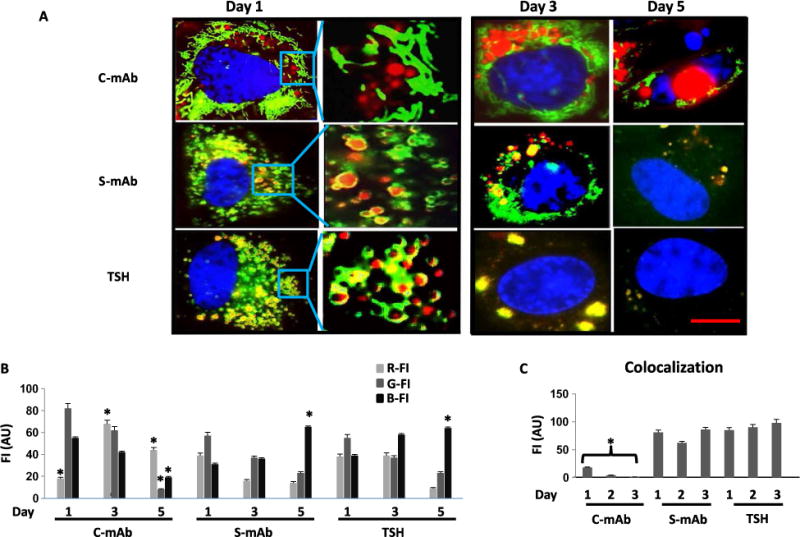
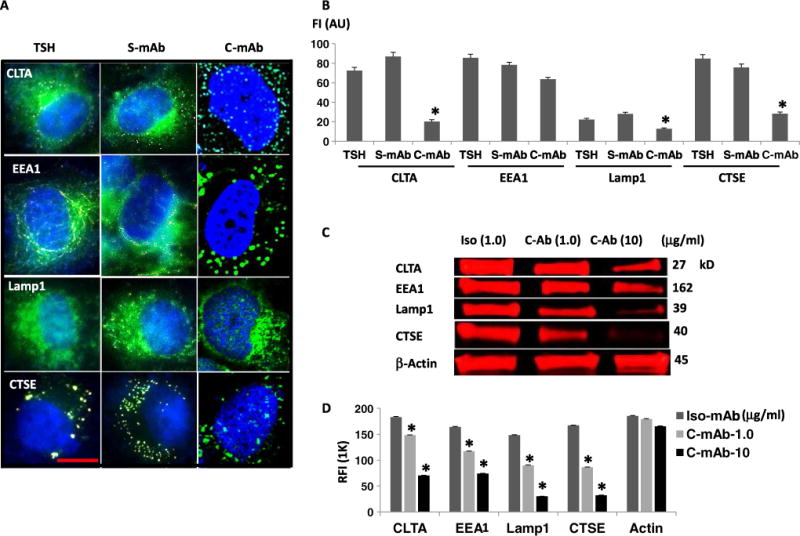
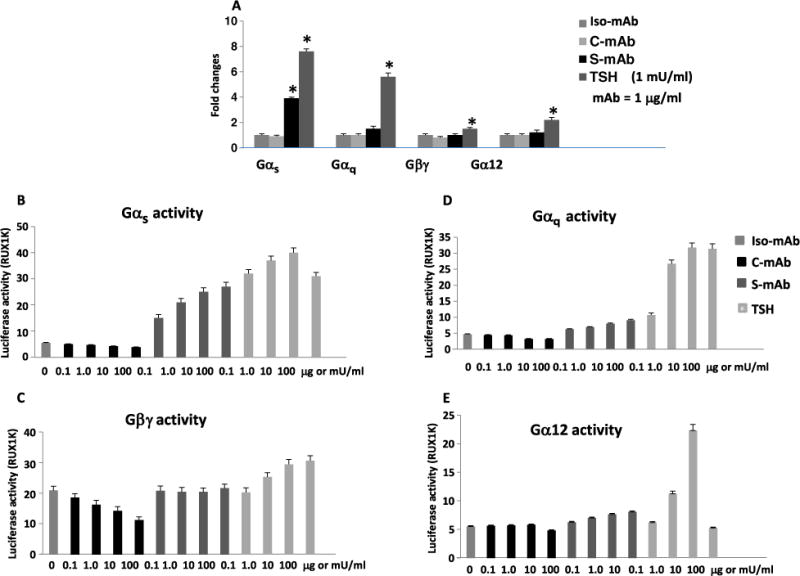
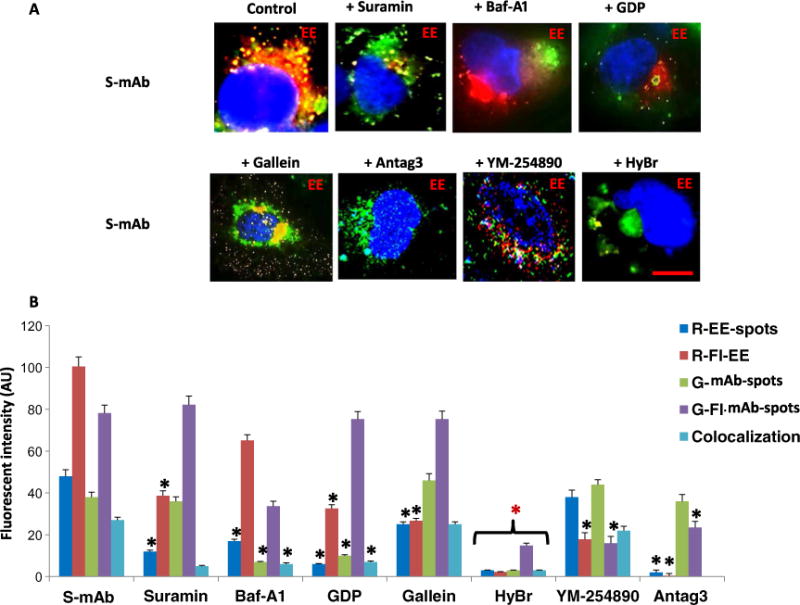
The thyroid-stimulating hormone receptor (TSHR) is a heterotrimeric guanine nucleotide-binding protein (G protein)-coupled receptor (GPCR). Autoimmune hyperthyroidism, commonly known as Graves' disease (GD), is caused by stimulating autoantibodies to the TSHR. We previously described TSHR-specific antibodies (TSHR-Abs) in GD that recognize linear epitopes in the cleavage region of the TSHR ectodomain (C-TSHR-Abs) and induce thyroid cell apoptosis instead of stimulating the TSHR. We found that C-TSHR-Abs entered the cell through clathrin-mediated endocytosis but did not trigger endosomal maturation and failed to undergo normal vesicular sorting and trafficking. We found that stimulating TSHR-Abs (S-TSHR-Abs) activated Gαs and, to a lesser extent, Gαq but that C-TSHR-Abs failed to activate any of the G proteins normally activated in response to TSH. Furthermore, specific inhibition of G proteins in the presence of S-TSHR-mAbs or TSH resulted in a similar failure of endosomal maturation as that caused by C-TSHR-mAbs. Hence, whereas S-TSHR-mAbs and TSH contributed to normal vesicular trafficking of TSHR through the activation of major G proteins, the C-TSHR-Abs resulted in GRK2- and β-arrestin-1-dependent biased signaling, which is interpreted as a danger signal by the cell. Our observations suggest that the binding of antibodies to different TSHR epitopes may decrease cell survival. Antibody-induced cell injury and the response to cell death amplify the loss of self-tolerance, which most likely helps to perpetuate GPCR-mediated autoimmunity.
Copyright © 2018 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Conflict of interest statement
Figures
References
-
- Li J, Ning Y, Hedley W, Saunders B, Chen Y, Tindill N, Hannay T, Subramaniam S. The Molecule Pages database. Nature. 2002;420:716–717. - PubMed
-
- Kobilka BK. Agonist-induced conformational changes in the β2 adrenergic receptor. J Pept Res. 2002;60:317–321. - PubMed
-
- Farid NR, Szkudlinski MW. Minireview: Structural and functional evolution of the thyrotropin receptor. Endocrinology. 2004;145:4048–4057. - PubMed
-
- Calebiro D, Nikolaev VO, Lohse MJ. Imaging of persistent cAMP signaling by internalized G protein-coupled receptors. J Mol Endocrinol. 2010;45:1–8. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
