

Evolutionary routes and KRAS dosage define pancreatic cancer phenotypes
- PMID: 29364867
- PMCID: PMC6097607
- DOI: 10.1038/nature25459
Evolutionary routes and KRAS dosage define pancreatic cancer phenotypes
Abstract
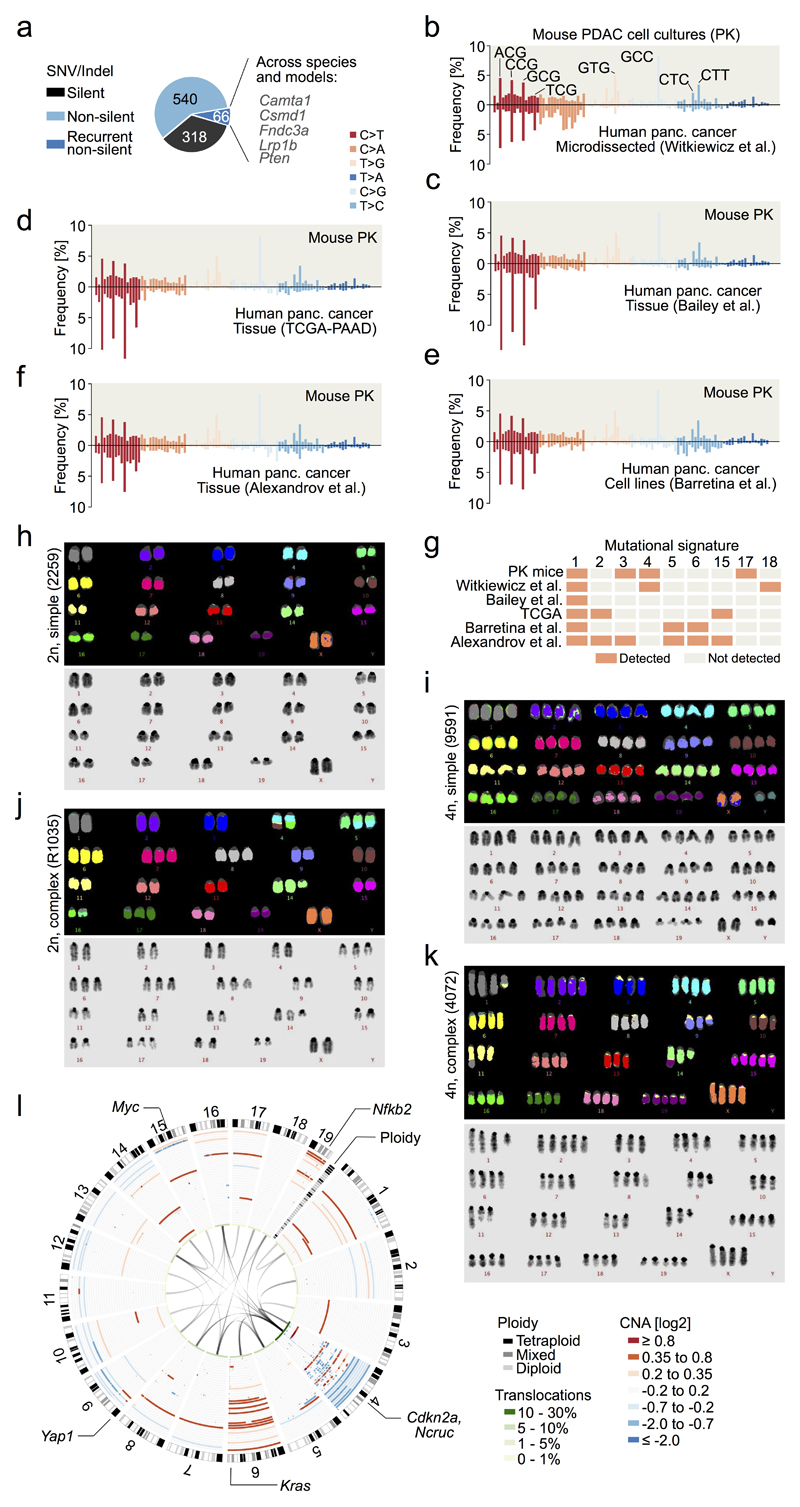
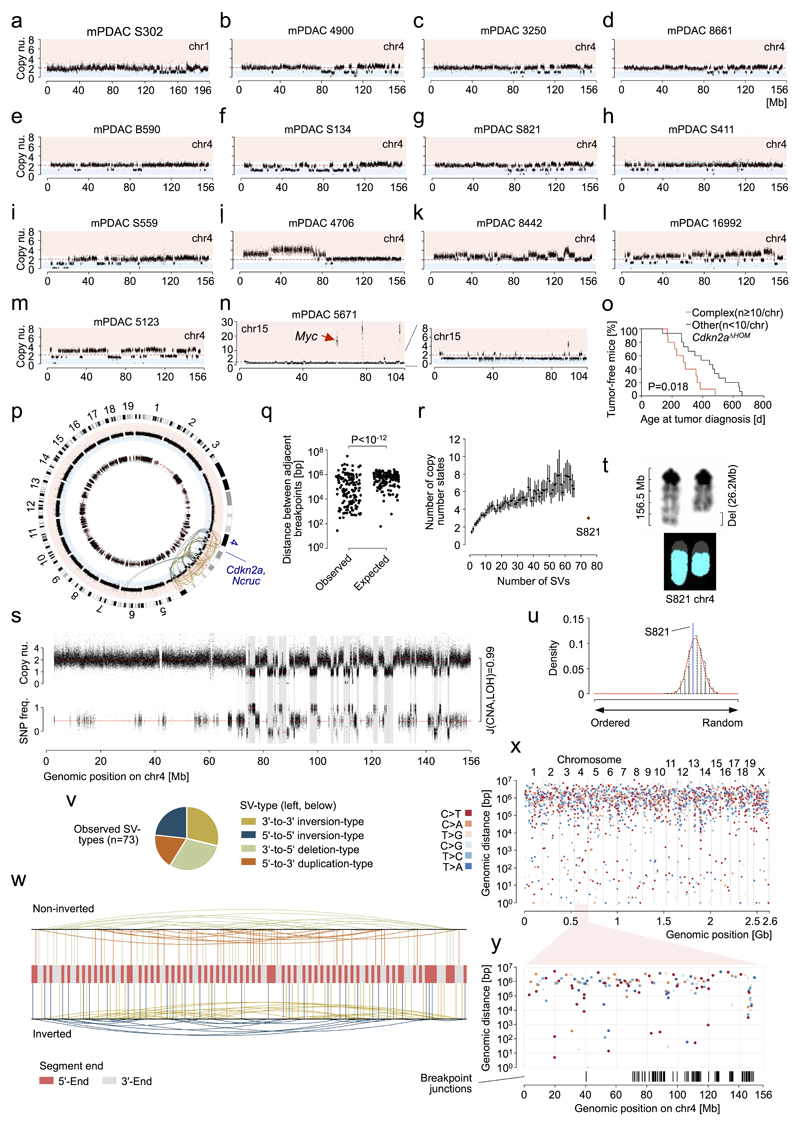
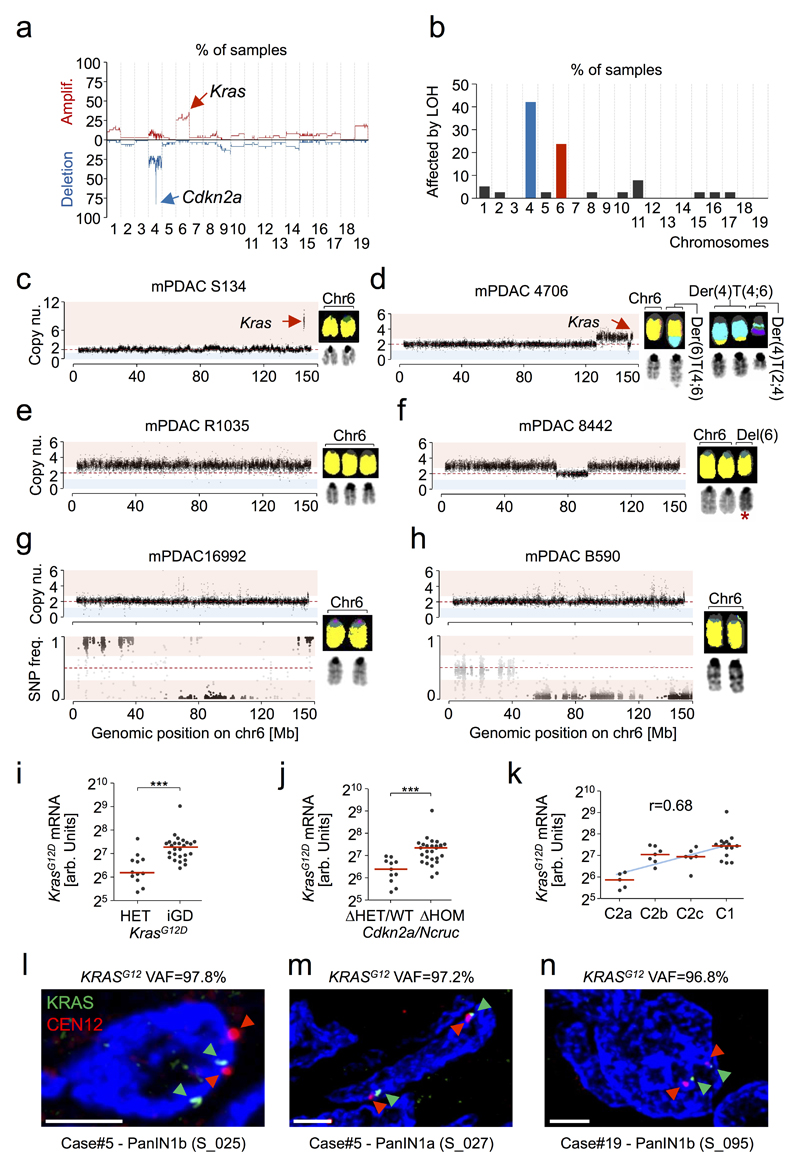
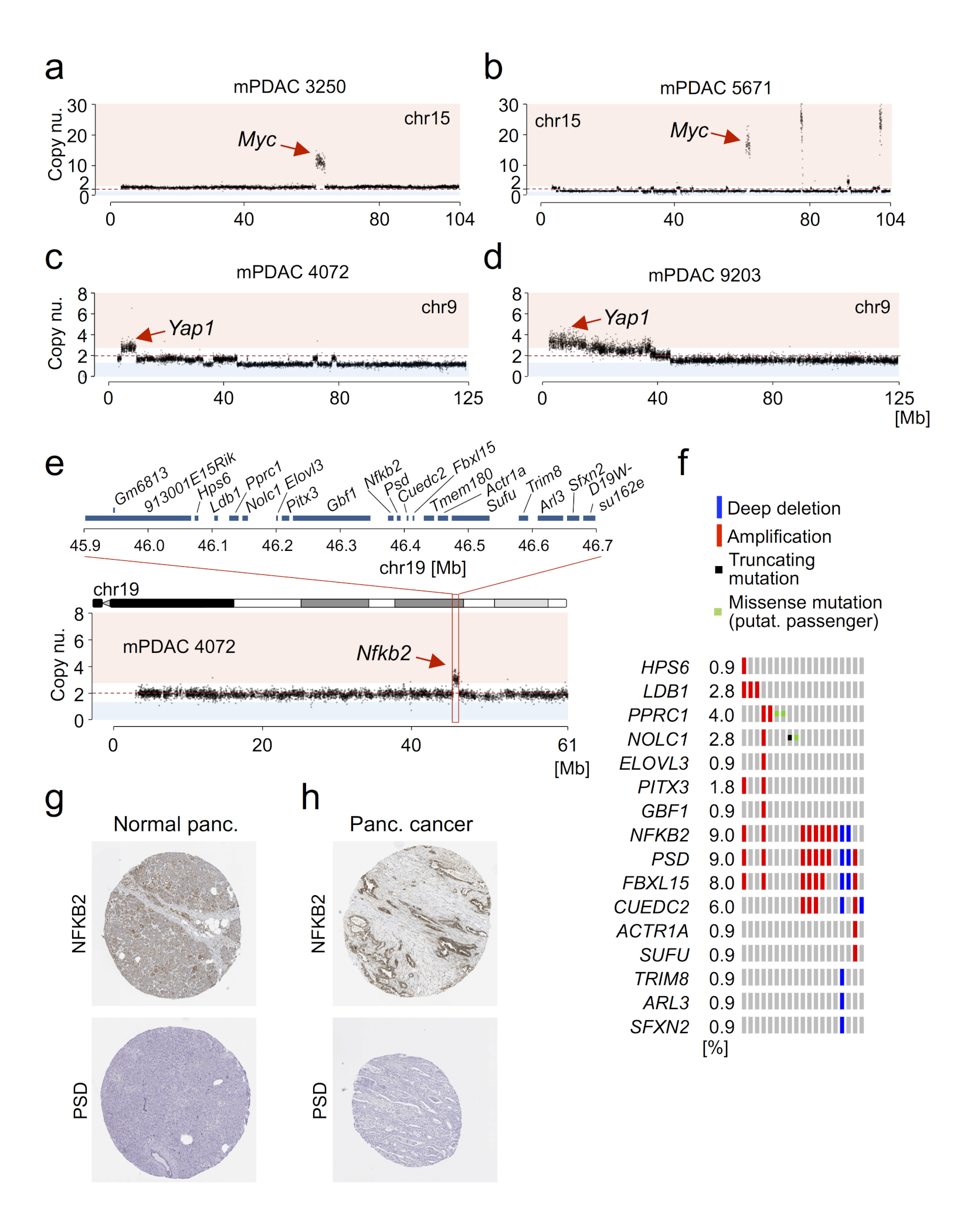
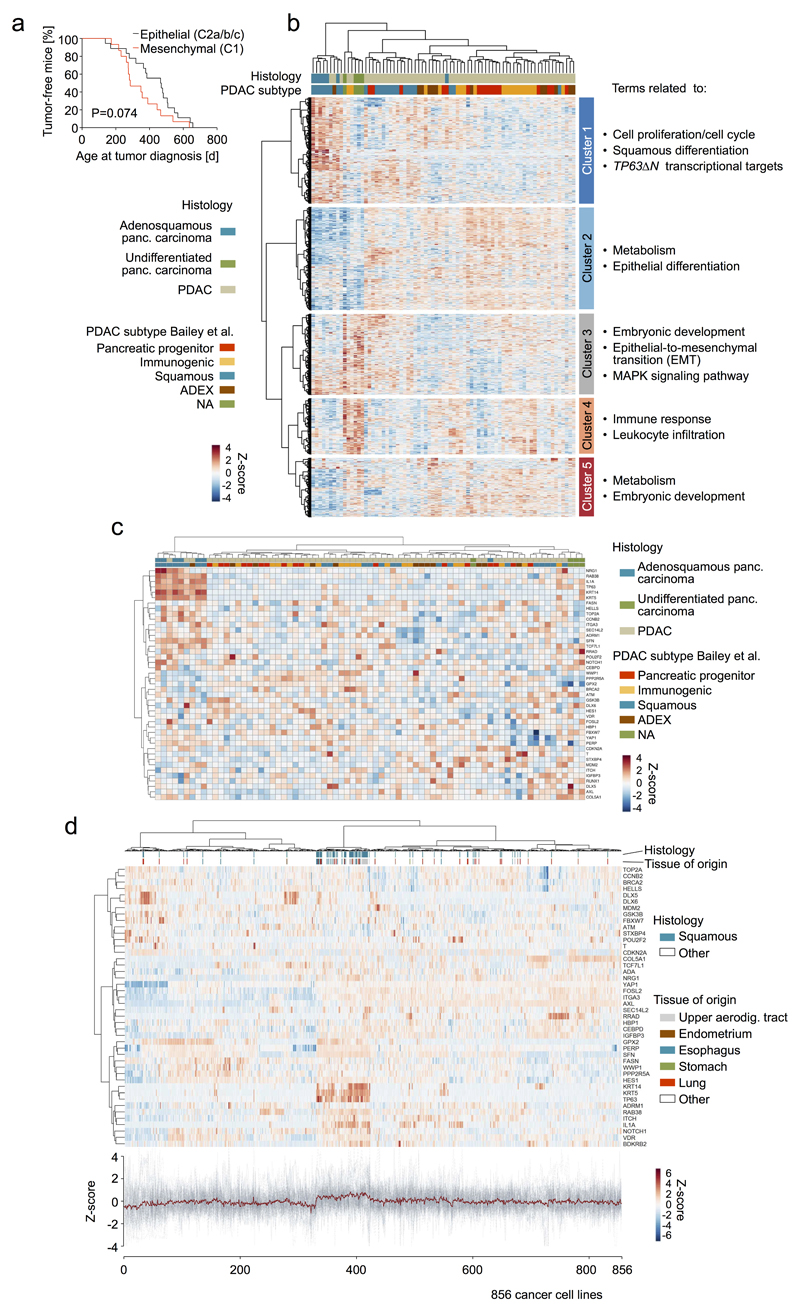
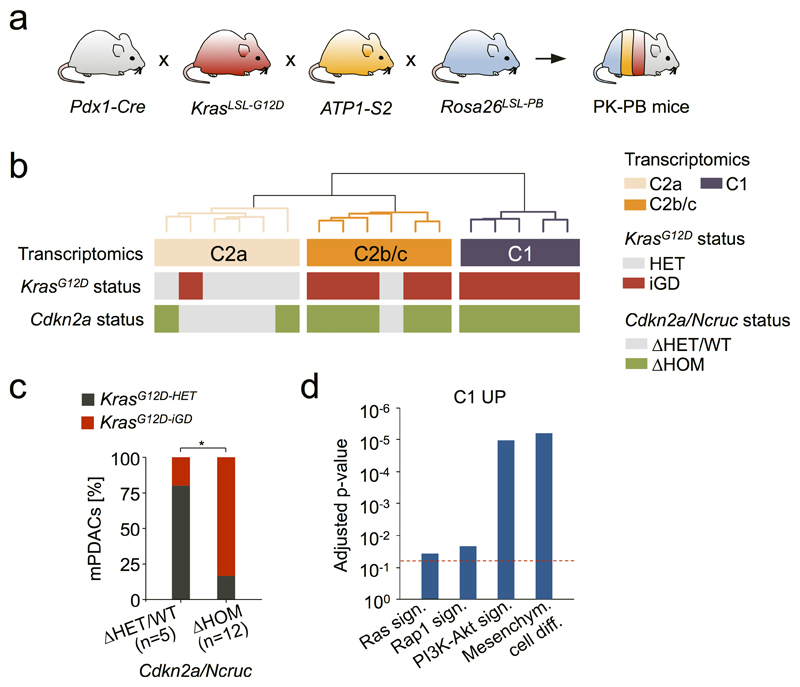
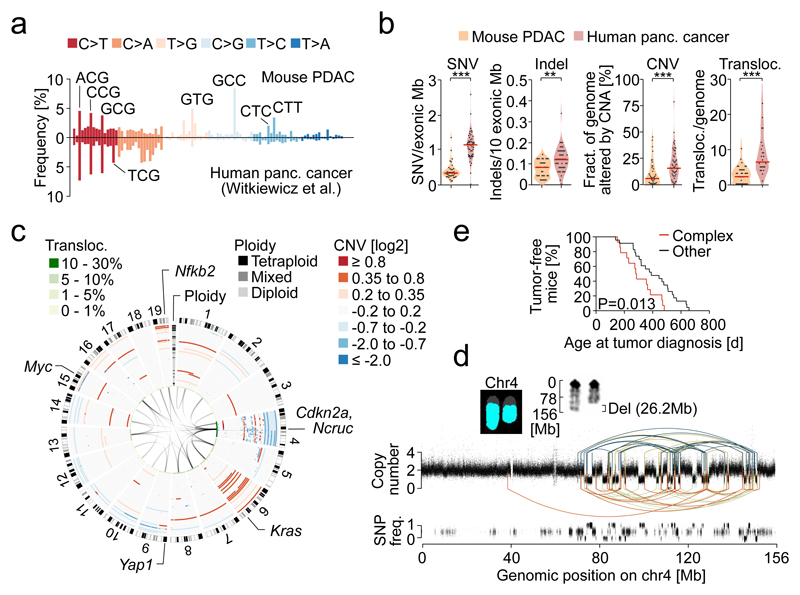
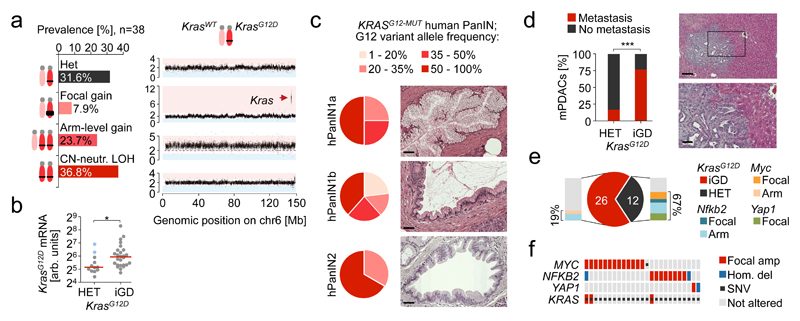
The poor correlation of mutational landscapes with phenotypes limits our understanding of the pathogenesis and metastasis of pancreatic ductal adenocarcinoma (PDAC). Here we show that oncogenic dosage-variation has a critical role in PDAC biology and phenotypic diversification. We find an increase in gene dosage of mutant KRAS in human PDAC precursors, which drives both early tumorigenesis and metastasis and thus rationalizes early PDAC dissemination. To overcome the limitations posed to gene dosage studies by the stromal richness of PDAC, we have developed large cell culture resources of metastatic mouse PDAC. Integration of cell culture genomes, transcriptomes and tumour phenotypes with functional studies and human data reveals additional widespread effects of oncogenic dosage variation on cell morphology and plasticity, histopathology and clinical outcome, with the highest KrasMUT levels underlying aggressive undifferentiated phenotypes. We also identify alternative oncogenic gains (Myc, Yap1 or Nfkb2), which collaborate with heterozygous KrasMUT in driving tumorigenesis, but have lower metastatic potential. Mechanistically, different oncogenic gains and dosages evolve along distinct evolutionary routes, licensed by defined allelic states and/or combinations of hallmark tumour suppressor alterations (Cdkn2a, Trp53, Tgfβ-pathway). Thus, evolutionary constraints and contingencies direct oncogenic dosage gain and variation along defined routes to drive the early progression of PDAC and shape its downstream biology. Our study uncovers universal principles of Ras-driven oncogenesis that have potential relevance beyond pancreatic cancer.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
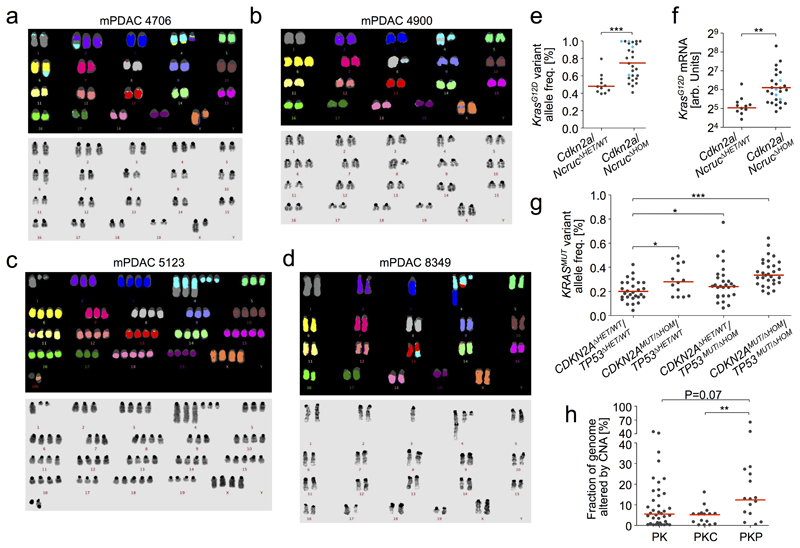
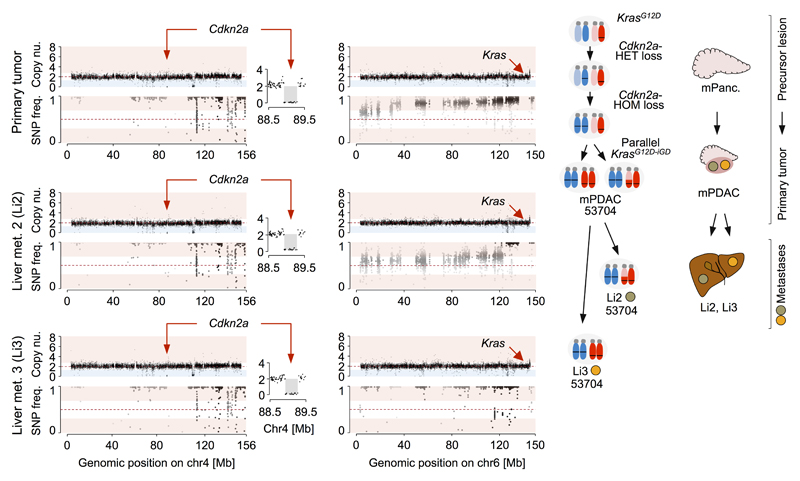
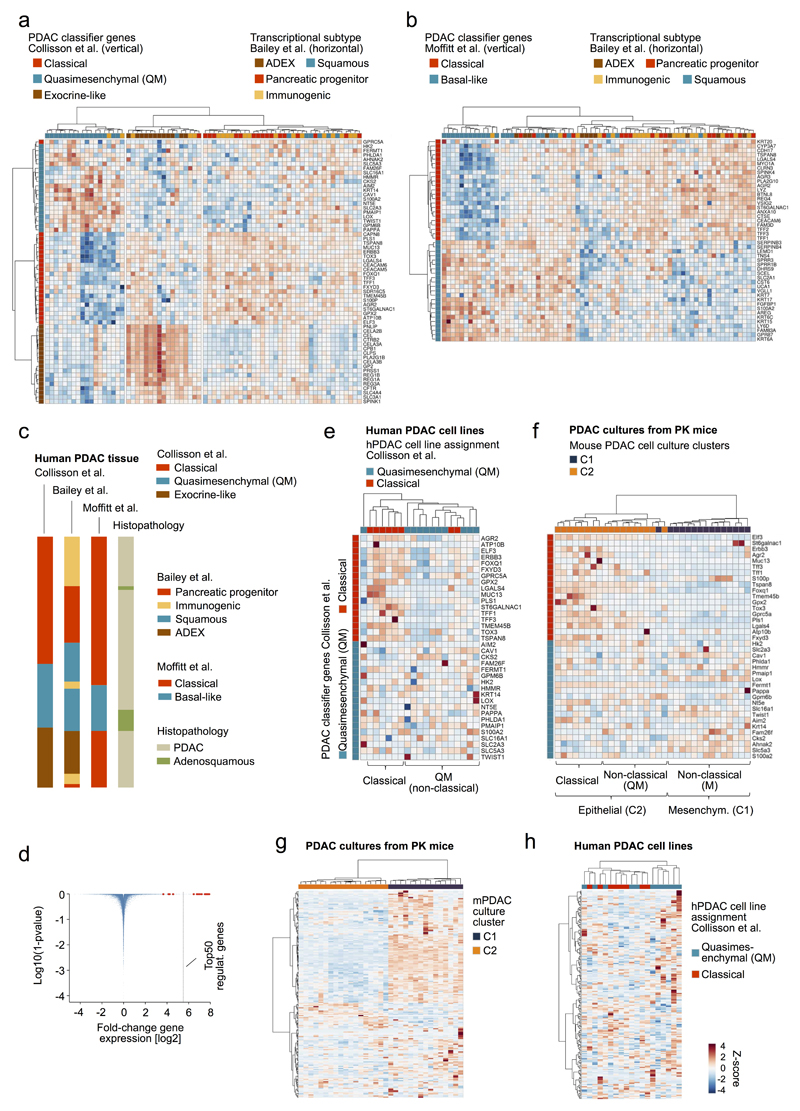
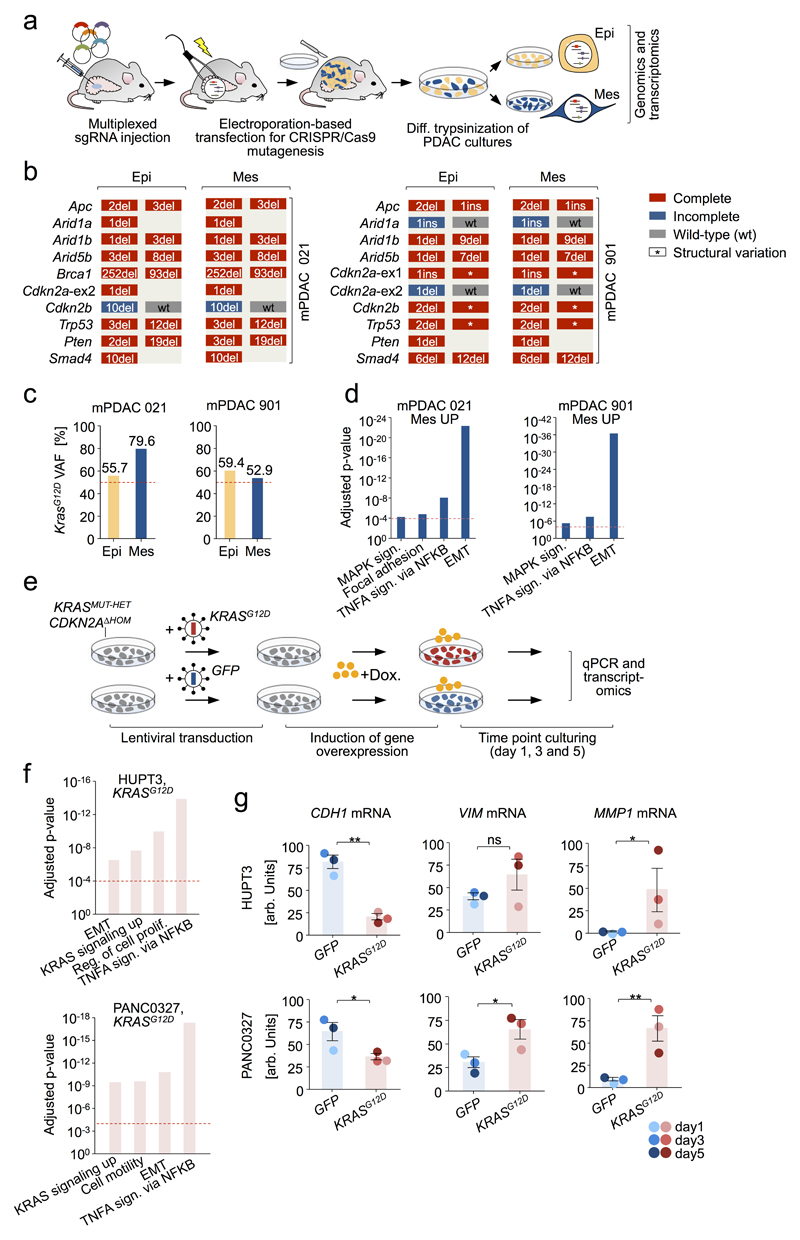
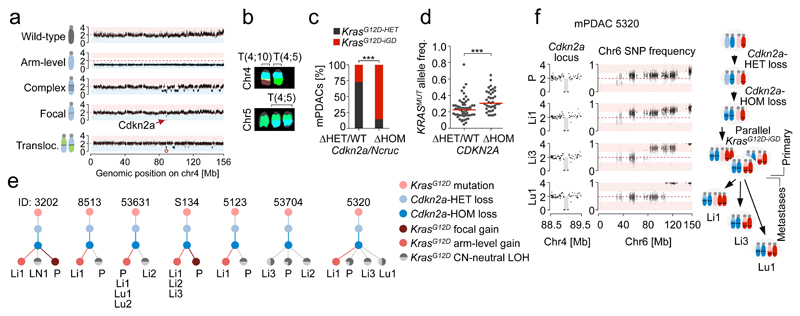
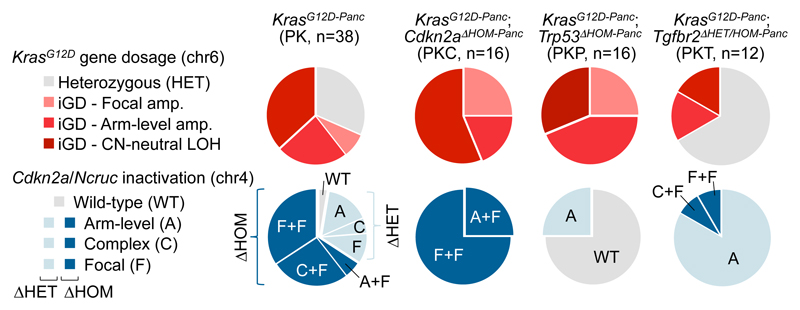
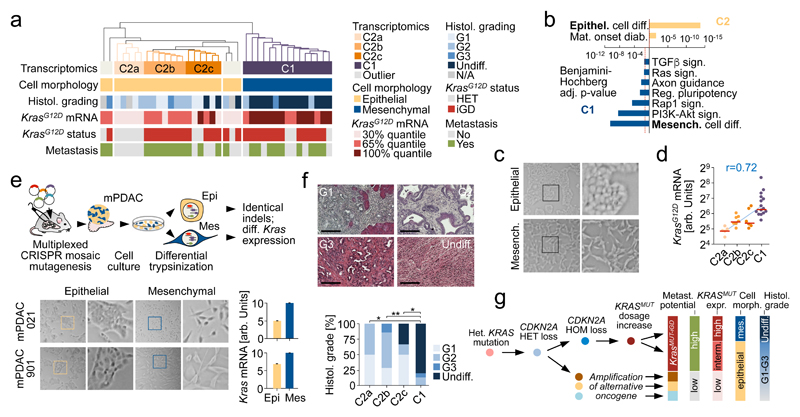
Comment in
-
KRASG12D Gene Dosage Drives Pancreatic Tumor Evolution and Progression.Cancer Discov. 2018 Mar;8(3):265. doi: 10.1158/2159-8290.CD-RW2018-020. Epub 2018 Feb 2. Cancer Discov. 2018. PMID: 29420182
-
Mutant Kras Dosage and Chromothripsis: The Right Ingredients for a Pancreatic Cancer Catastrophe.Trends Cancer. 2018 Jun;4(6):399-401. doi: 10.1016/j.trecan.2018.04.004. Epub 2018 May 3. Trends Cancer. 2018. PMID: 29860982
-
Increased mutant KRAS gene dosage drives pancreatic cancer progression: evidence for wild-type KRAS as a tumor suppressor?Hepatobiliary Surg Nutr. 2018 Oct;7(5):403-405. doi: 10.21037/hbsn.2018.07.03. Hepatobiliary Surg Nutr. 2018. PMID: 30498719 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
