

Non-alcoholic steatohepatitis pathogenesis: sublethal hepatocyte injury as a driver of liver inflammation
- PMID: 29367207
- PMCID: PMC5889737
- DOI: 10.1136/gutjnl-2017-315691
Non-alcoholic steatohepatitis pathogenesis: sublethal hepatocyte injury as a driver of liver inflammation
Abstract
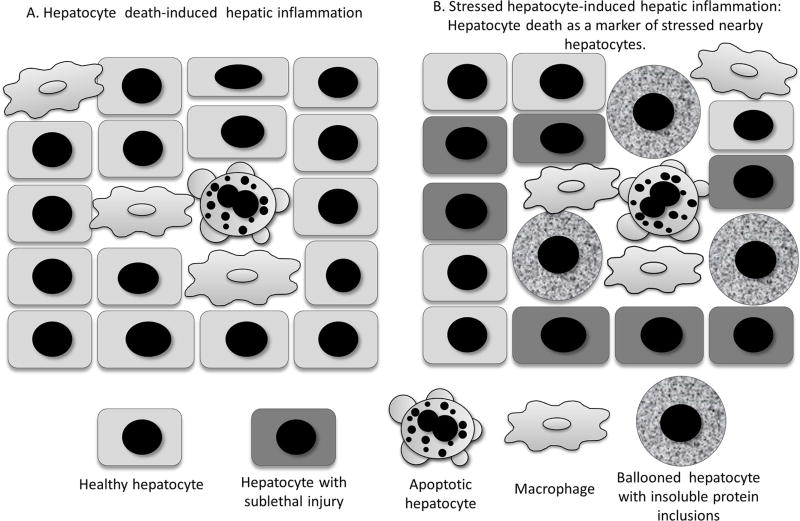
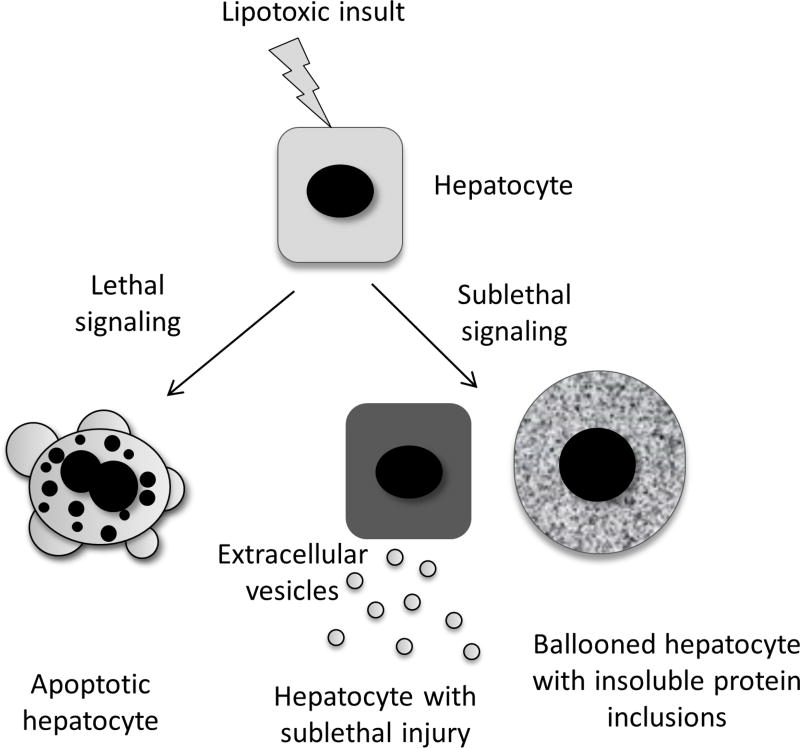
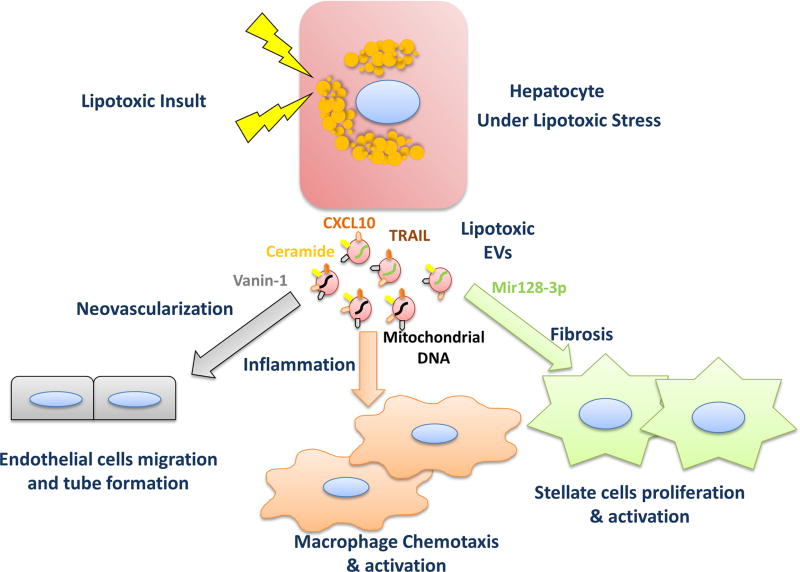
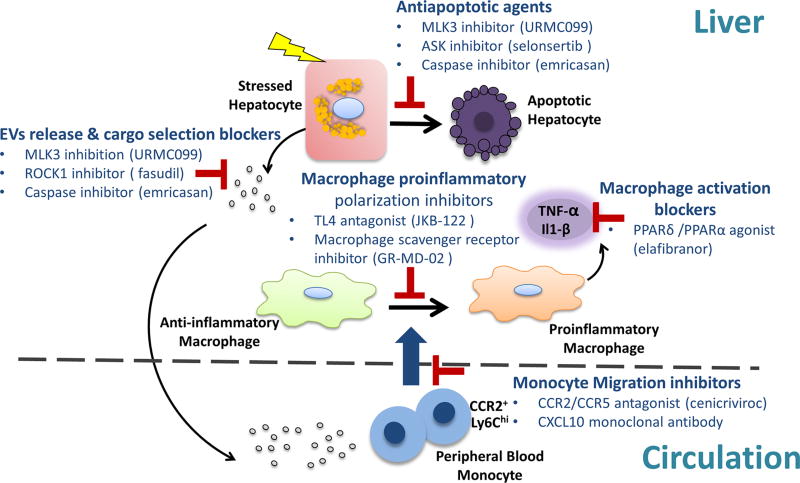
A subset of patients with non-alcoholic fatty liver disease develop an inflammatory condition, termed non-alcoholic steatohepatitis (NASH). NASH is characterised by hepatocellular injury, innate immune cell-mediated inflammation and progressive liver fibrosis. The mechanisms whereby hepatic inflammation occurs in NASH remain incompletely understood, but appear to be linked to the proinflammatory microenvironment created by toxic lipid-induced hepatocyte injury, termed lipotoxicity. In this review, we discuss the signalling pathways induced by sublethal hepatocyte lipid overload that contribute to the pathogenesis of NASH. Furthermore, we will review the role of proinflammatory, proangiogenic and profibrotic hepatocyte-derived extracellular vesicles as disease biomarkers and pathogenic mediators during lipotoxicity. We also review the potential therapeutic strategies to block the feed-forward loop between sublethal hepatocyte injury and liver inflammation.
Keywords: inflammation; nonalcoholic steatohepatitis.
© Article author(s) (or their employer(s) unless otherwise stated in the text of the article) 2018. All rights reserved. No commercial use is permitted unless otherwise expressly granted.
Conflict of interest statement
Competing interests: None declared.
Figures
References
-
- Younossi ZM, Koenig AB, Abdelatif D, et al. Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64:73–84. - PubMed
-
- Ahmed A, Wong RJ, Harrison SA. Nonalcoholic fatty liver disease review: diagnosis, treatment, and outcomes. Clin Gastroenterol Hepatol. 2015;13:2062–70. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical