

IL-36/LXR axis modulates cholesterol metabolism and immune defense to Mycobacterium tuberculosis
- PMID: 29367626
- PMCID: PMC5784124
- DOI: 10.1038/s41598-018-19476-x
IL-36/LXR axis modulates cholesterol metabolism and immune defense to Mycobacterium tuberculosis
Abstract
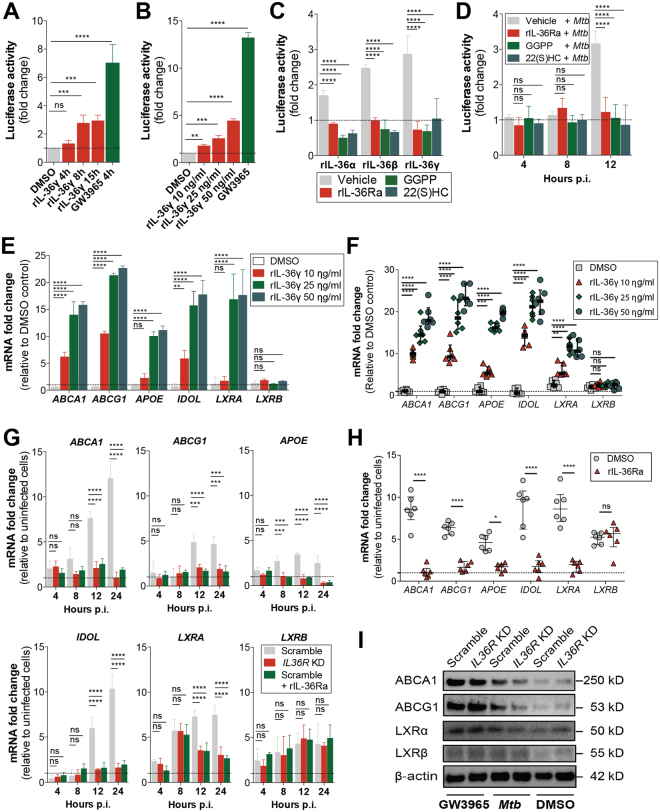
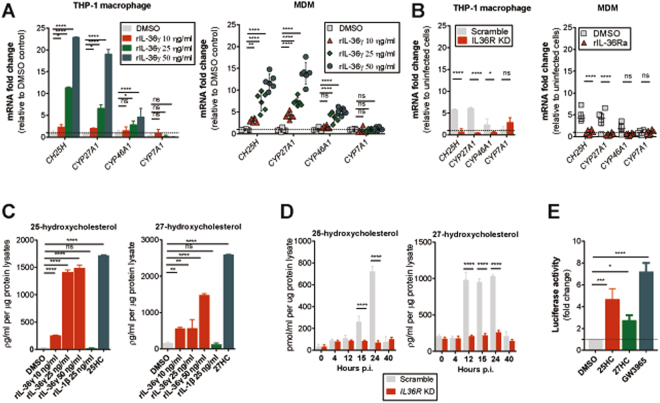
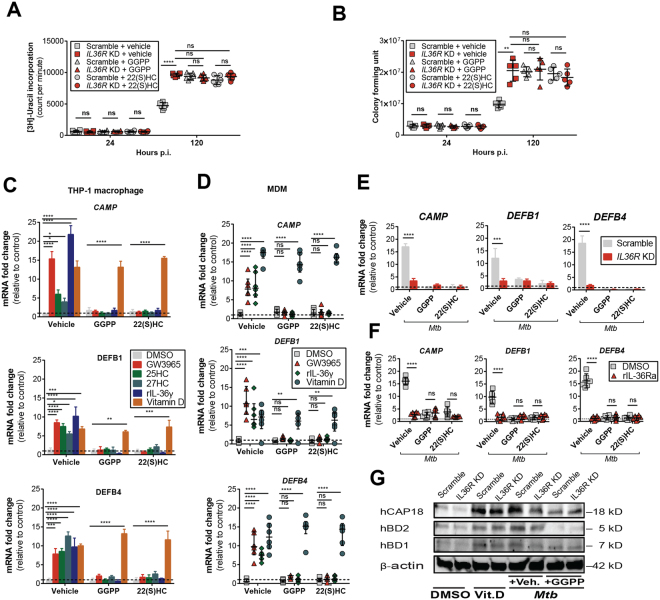
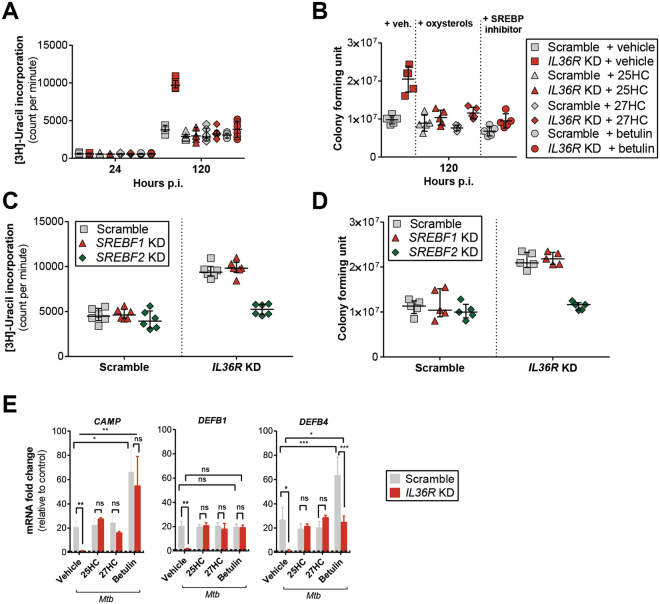
Mycobacterium tuberculosis (Mtb) is a life-threatening pathogen in humans. Bacterial infection of macrophages usually triggers strong innate immune mechanisms, including IL-1 cytokine secretion. The newer member of the IL-1 family, IL-36, was recently shown to be involved in cellular defense against Mtb. To unveil the underlying mechanism of IL-36 induced antibacterial activity, we analyzed its role in the regulation of cholesterol metabolism, together with the involvement of Liver X Receptor (LXR) in this process. We report that, in Mtb-infected macrophages, IL-36 signaling modulates cholesterol biosynthesis and efflux via LXR. Moreover, IL-36 induces the expression of cholesterol-converting enzymes and the accumulation of LXR ligands, such as oxysterols. Ultimately, both IL-36 and LXR signaling play a role in the regulation of antimicrobial peptides expression and in Mtb growth restriction. These data provide novel evidence for the importance of IL-36 and cholesterol metabolism mediated by LXR in cellular host defense against Mtb.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures


References
-
- WHO. Global tuberculosis report. (World Health Organization, Geneva, Switzerland, 2017).
-
- Ernst, J. D. The immunological life cycle of tuberculosis. Nature reviews Immunology12, 581–591, 10.1038/nri3259 (2012). - PubMed
-
- Dorhoi, A. & Kaufmann, S. H. Pathology and immune reactivity: understanding multidimensionality in pulmonary tuberculosis. Seminars in immunopathology, 10.1007/s00281-015-0531-3 (2015). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
