

CardioClassifier: disease- and gene-specific computational decision support for clinical genome interpretation
- PMID: 29369293
- PMCID: PMC6558251
- DOI: 10.1038/gim.2017.258
CardioClassifier: disease- and gene-specific computational decision support for clinical genome interpretation
Abstract
Purpose: Internationally adopted variant interpretation guidelines from the American College of Medical Genetics and Genomics (ACMG) are generic and require disease-specific refinement. Here we developed CardioClassifier ( http://www.cardioclassifier.org ), a semiautomated decision-support tool for inherited cardiac conditions (ICCs).
Methods: CardioClassifier integrates data retrieved from multiple sources with user-input case-specific information, through an interactive interface, to support variant interpretation. Combining disease- and gene-specific knowledge with variant observations in large cohorts of cases and controls, we refined 14 computational ACMG criteria and created three ICC-specific rules.
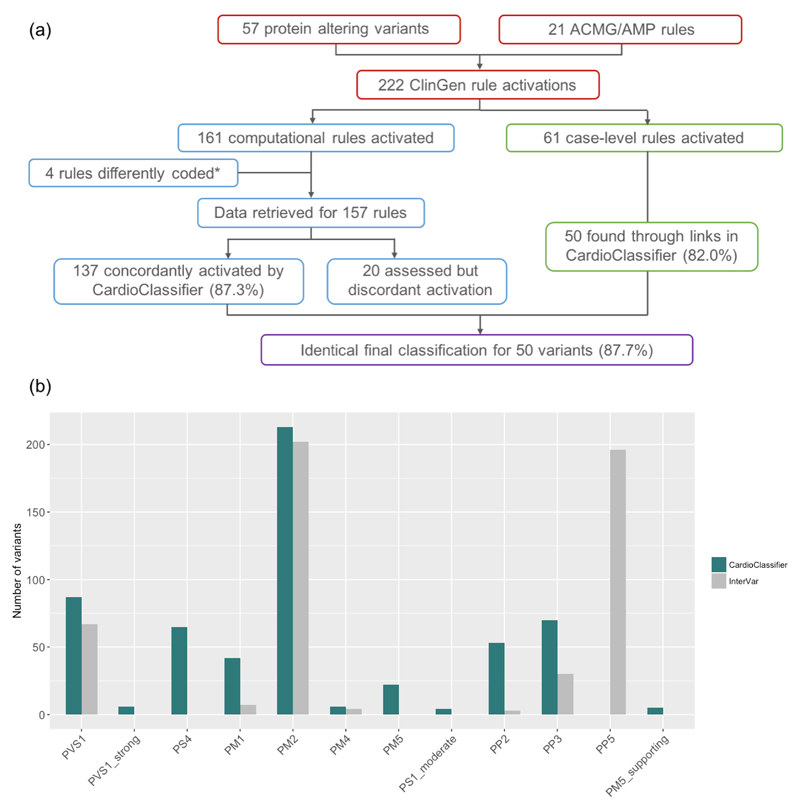
Results: We benchmarked CardioClassifier on 57 expertly curated variants and show full retrieval of all computational data, concordantly activating 87.3% of rules. A generic annotation tool identified fewer than half as many clinically actionable variants (64/219 vs. 156/219, Fisher's P = 1.1 × 10-18), with important false positives, illustrating the critical importance of disease and gene-specific annotations. CardioClassifier identified putatively disease-causing variants in 33.7% of 327 cardiomyopathy cases, comparable with leading ICC laboratories. Through addition of manually curated data, variants found in over 40% of cardiomyopathy cases are fully annotated, without requiring additional user-input data.
Conclusion: CardioClassifier is an ICC-specific decision-support tool that integrates expertly curated computational annotations with case-specific data to generate fast, reproducible, and interactive variant pathogenicity reports, according to best practice guidelines.
Keywords: bioinformatics; clinical genomics; inherited cardiac conditions; next-generation sequencing; variant interpretation.
Conflict of interest statement
Figures
References
-
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genetics in Medicine. 2015;17(5):405–423. doi: 10.1038/gim.2015.30. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
