

Extended FTLD pedigree segregating a Belgian GRN-null mutation: neuropathological heterogeneity in one family
- PMID: 29370838
- PMCID: PMC6389176
- DOI: 10.1186/s13195-017-0334-y
Extended FTLD pedigree segregating a Belgian GRN-null mutation: neuropathological heterogeneity in one family
Abstract
Background: In this paper, we describe the clinical and neuropathological findings of nine members of the Belgian progranulin gene (GRN) founder family. In this family, the loss-of-function mutation IVS1 + 5G > C was identified in 2006. In 2007, a clinical description of the mutation carriers was published that revealed the clinical heterogeneity among IVS1 + 5G > C carriers. We report our comparison of our data with the published clinical and neuropathological characteristics of other GRN mutations as well as other frontotemporal lobar degeneration (FTLD) syndromes, and we present a review of the literature.
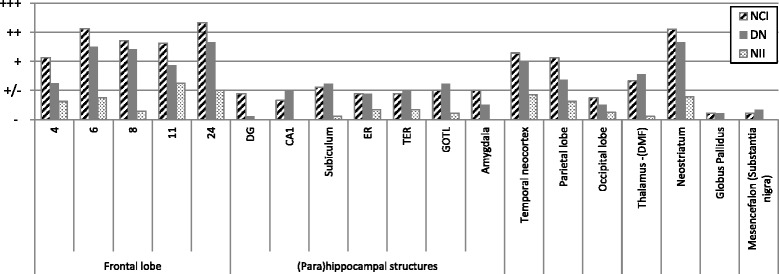
Methods: For each case, standardized sampling and staining were performed to identify proteinopathies, cerebrovascular disease, and hippocampal sclerosis.
Results: The neuropathological substrate in the studied family was compatible in all cases with transactive response DNA-binding protein (TDP) proteinopathy type A, as expected. Additionally, most of the cases presented also with primary age-related tauopathy (PART) or mild Alzheimer's disease (AD) neuropathological changes, and one case had extensive Lewy body pathology. An additional finding was the presence of cerebral small vessel changes in every patient in this family.
Conclusions: Our data show not only that the IVS1 + 5G > C mutation has an exclusive association with FTLD-TDP type A proteinopathy but also that other proteinopathies can occur and should be looked for. Because the penetrance rate of the clinical phenotype of carriers of GRN mutations is age-dependent, further research is required to investigate the role of co-occurring age-related pathologies such as AD, PART, and cerebral small vessel disease.
Keywords: Cerebral small vessel disease (SVD); FTD; FTD-GRN; FTLD; FTLD-TDP; Frontotemporal dementia; Frontotemporal lobar degeneration.
Conflict of interest statement
Consent for publication
The patients and/or their legal representatives gave their consent for publication.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
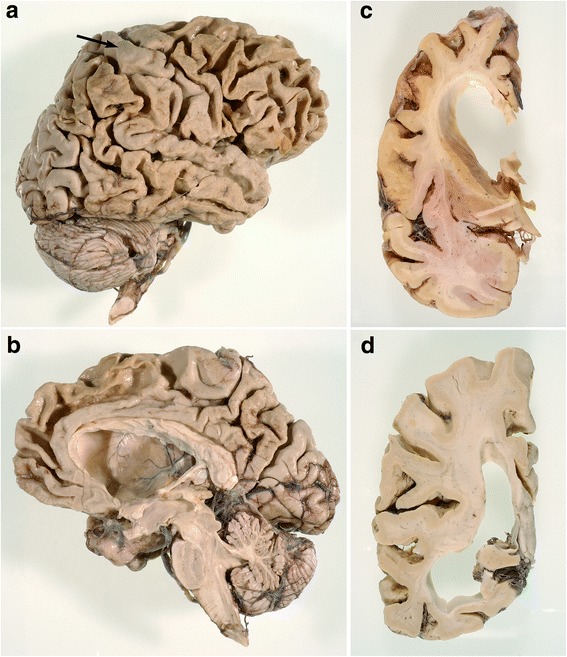
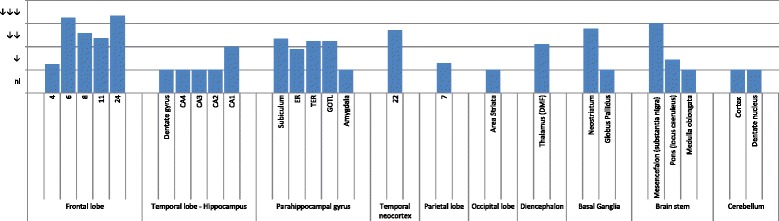
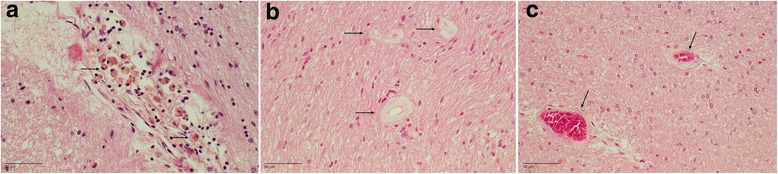
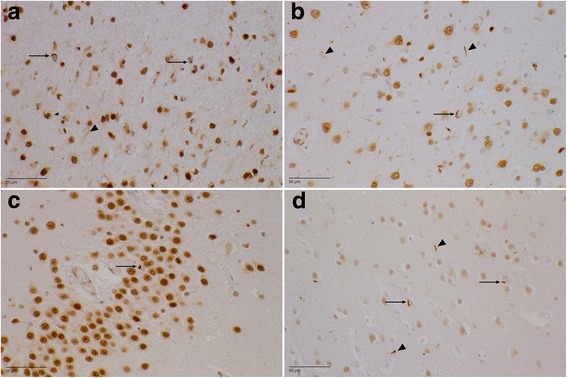
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
