

A global ocean atlas of eukaryotic genes
- PMID: 29371626
- PMCID: PMC5785536
- DOI: 10.1038/s41467-017-02342-1
A global ocean atlas of eukaryotic genes
Abstract
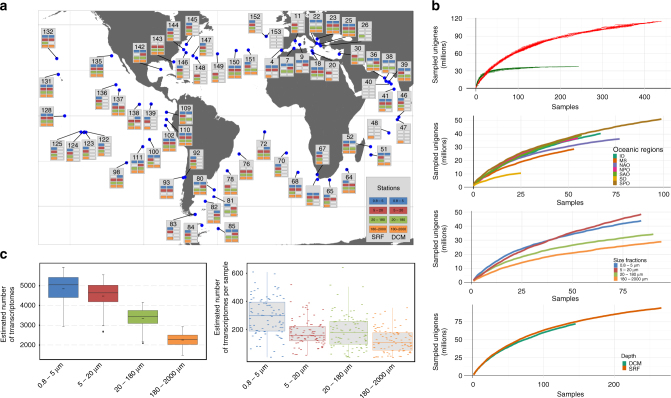
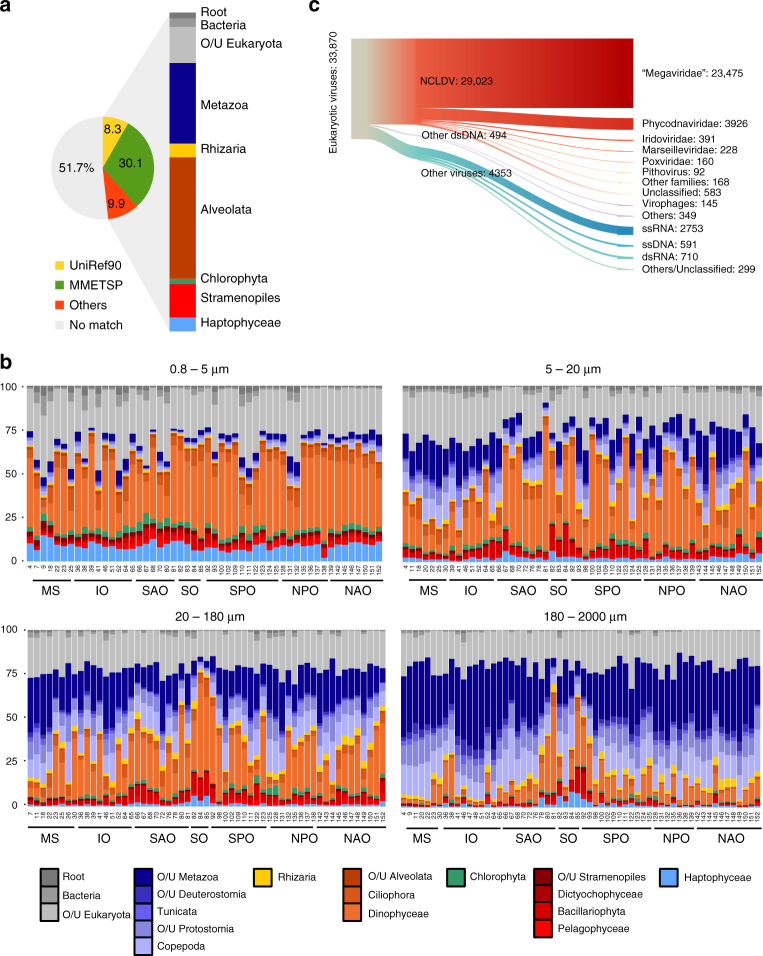
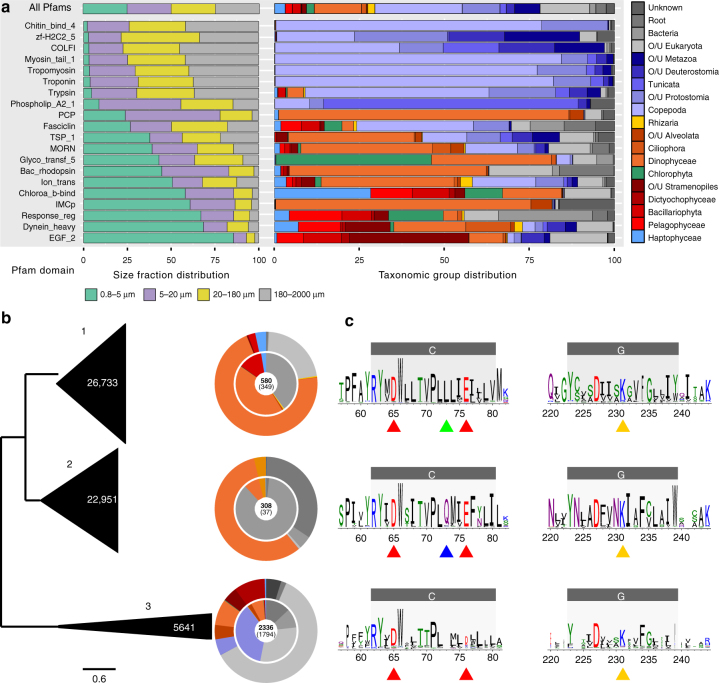
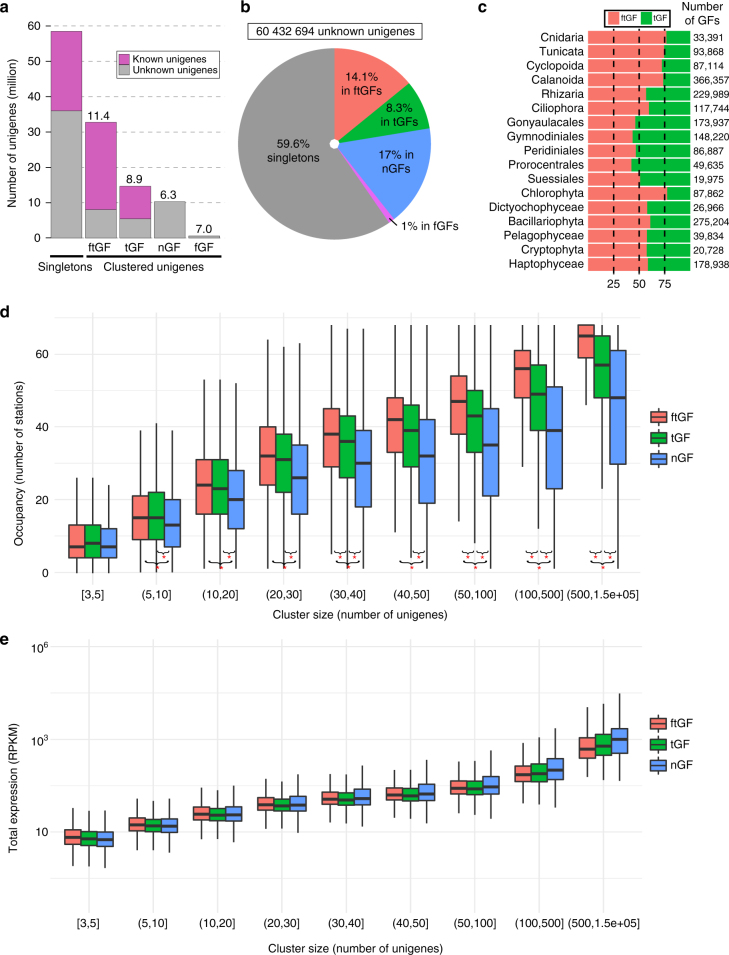
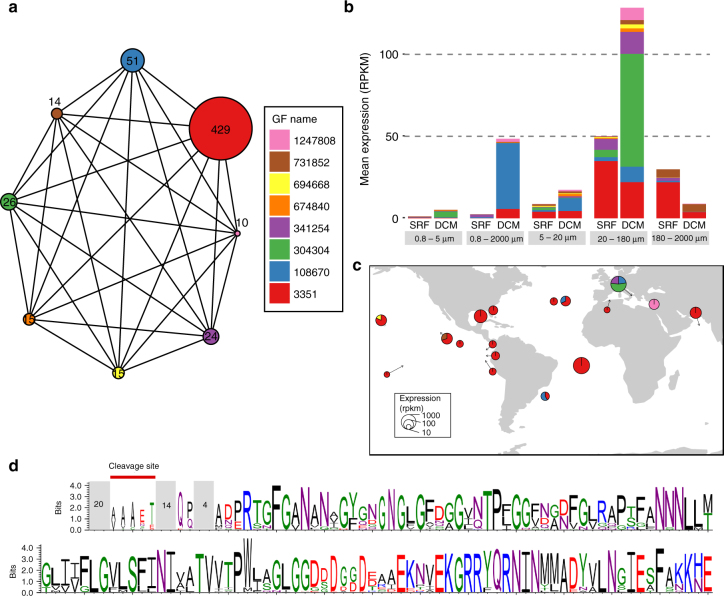
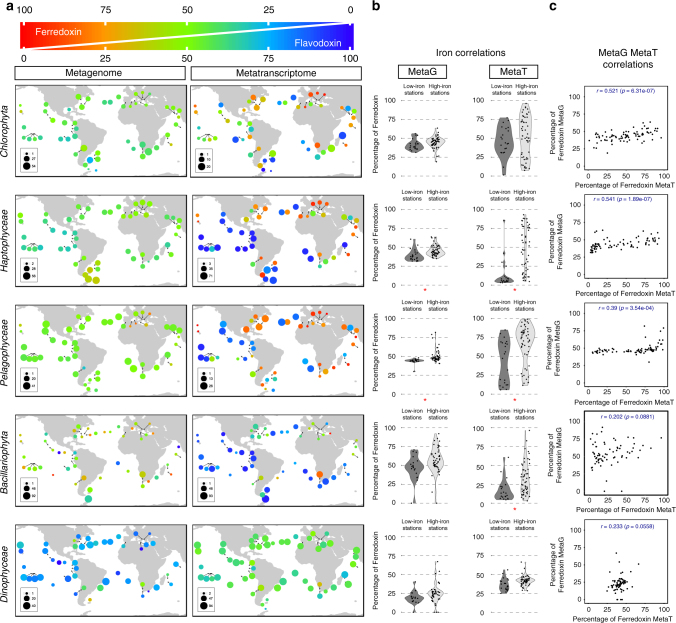
While our knowledge about the roles of microbes and viruses in the ocean has increased tremendously due to recent advances in genomics and metagenomics, research on marine microbial eukaryotes and zooplankton has benefited much less from these new technologies because of their larger genomes, their enormous diversity, and largely unexplored physiologies. Here, we use a metatranscriptomics approach to capture expressed genes in open ocean Tara Oceans stations across four organismal size fractions. The individual sequence reads cluster into 116 million unigenes representing the largest reference collection of eukaryotic transcripts from any single biome. The catalog is used to unveil functions expressed by eukaryotic marine plankton, and to assess their functional biogeography. Almost half of the sequences have no similarity with known proteins, and a great number belong to new gene families with a restricted distribution in the ocean. Overall, the resource provides the foundations for exploring the roles of marine eukaryotes in ocean ecology and biogeochemistry.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Dortch Q, Packard T. Differences in biomass structure between oligotrophic and eutrophic marine ecosystems. Deep Sea Res. 1989;36:223–240. doi: 10.1016/0198-0149(89)90135-0. - DOI
-
- Gasol JM, Giorgio PAD, Duarte CM. Biomass distribution in marine planktonic communities. Limnol. Oceanogr. 1997;42:1353–1363. doi: 10.4319/lo.1997.42.6.1353. - DOI
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
