

A broad atlas of somatic hypermutation allows prediction of activation-induced deaminase targets
- PMID: 29374026
- PMCID: PMC5839764
- DOI: 10.1084/jem.20171738
A broad atlas of somatic hypermutation allows prediction of activation-induced deaminase targets
Abstract
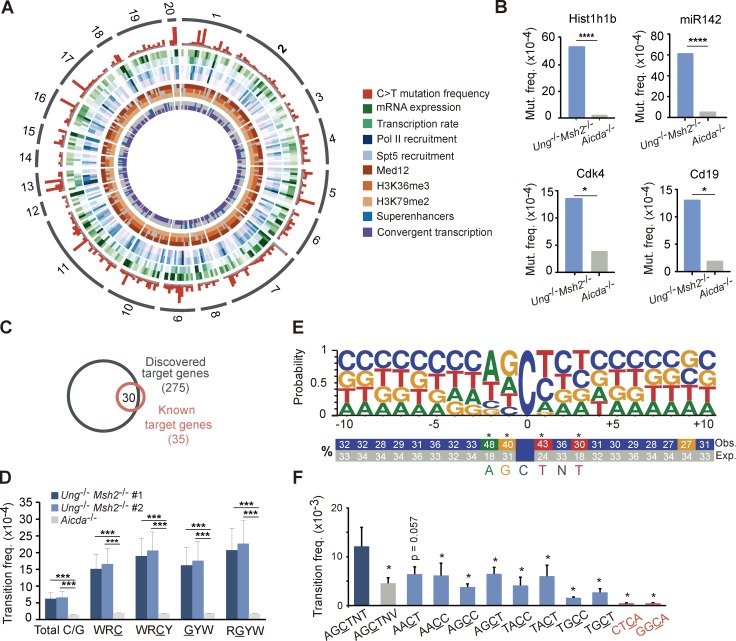
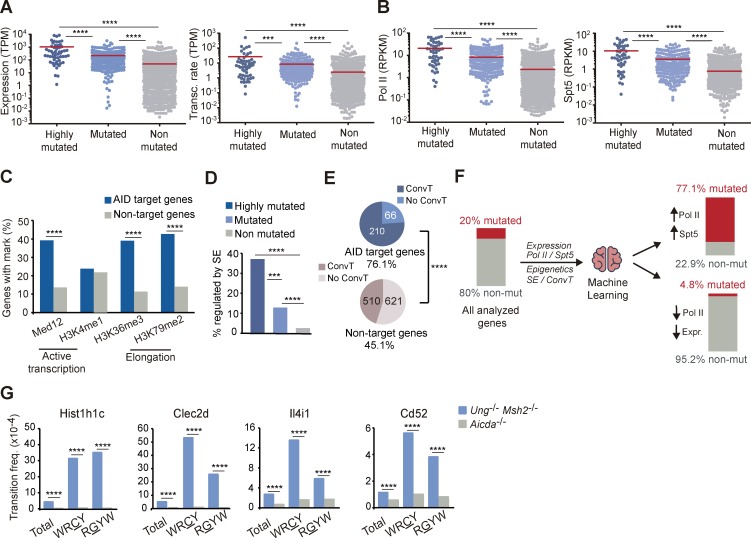
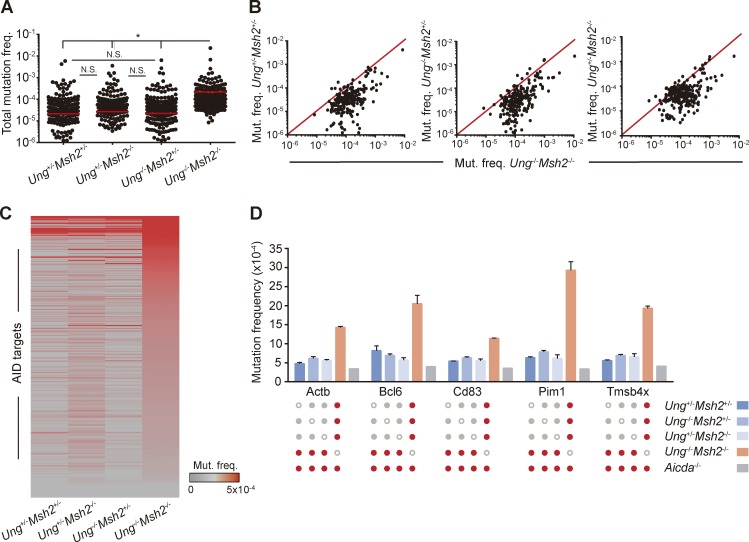
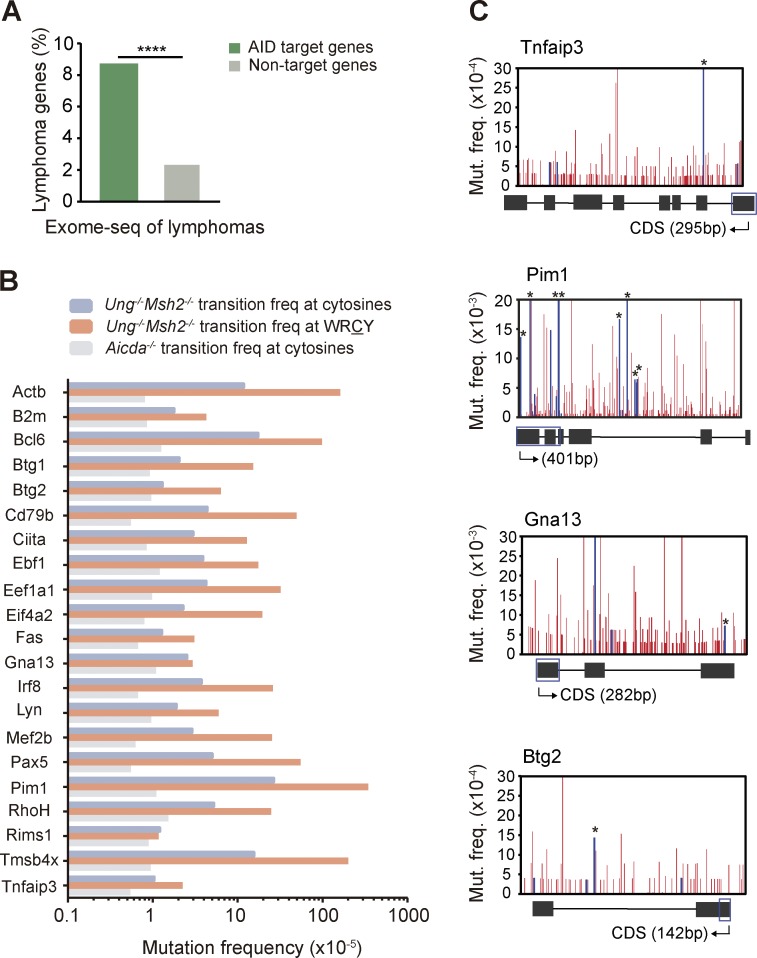
Activation-induced deaminase (AID) initiates antibody diversification in germinal center (GC) B cells through the deamination of cytosines on immunoglobulin genes. AID can also target other regions in the genome, triggering mutations or chromosome translocations, with major implications for oncogenic transformation. However, understanding the specificity of AID has proved extremely challenging. We have sequenced at very high depth >1,500 genomic regions from GC B cells and identified 275 genes targeted by AID, including 30 of the previously known 35 AID targets. We have also identified the most highly mutated hotspot for AID activity described to date. Furthermore, integrative analysis of the molecular features of mutated genes coupled to machine learning has produced a powerful predictive tool for AID targets. We also have found that base excision repair and mismatch repair back up each other to faithfully repair AID-induced lesions. Finally, our data establish a novel link between AID mutagenic activity and lymphomagenesis.
© 2018 Álvarez-Prado et al.
Figures
Comment in
-
Predicting AID off-targets: A step forward.J Exp Med. 2018 Mar 5;215(3):721-722. doi: 10.1084/jem.20180231. Epub 2018 Feb 15. J Exp Med. 2018. PMID: 29449308 Free PMC article.
References
-
- Chiarle R., Zhang Y., Frock R.L., Lewis S.M., Molinie B., Ho Y.J., Myers D.R., Choi V.W., Compagno M., Malkin D.J., et al. 2011. Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell. 147:107–119. 10.1016/j.cell.2011.07.049 - DOI - PMC - PubMed
-
- de Miranda N.F., Georgiou K., Chen L., Wu C., Gao Z., Zaravinos A., Lisboa S., Enblad G., Teixeira M.R., Zeng Y., et al. 2014. Exome sequencing reveals novel mutation targets in diffuse large B-cell lymphomas derived from Chinese patients. Blood. 124:2544–2553. 10.1182/blood-2013-12-546309 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
