

Hypothalamic loss of Snord116 recapitulates the hyperphagia of Prader-Willi syndrome
- PMID: 29376887
- PMCID: PMC5824864
- DOI: 10.1172/JCI97007
Hypothalamic loss of Snord116 recapitulates the hyperphagia of Prader-Willi syndrome
Abstract
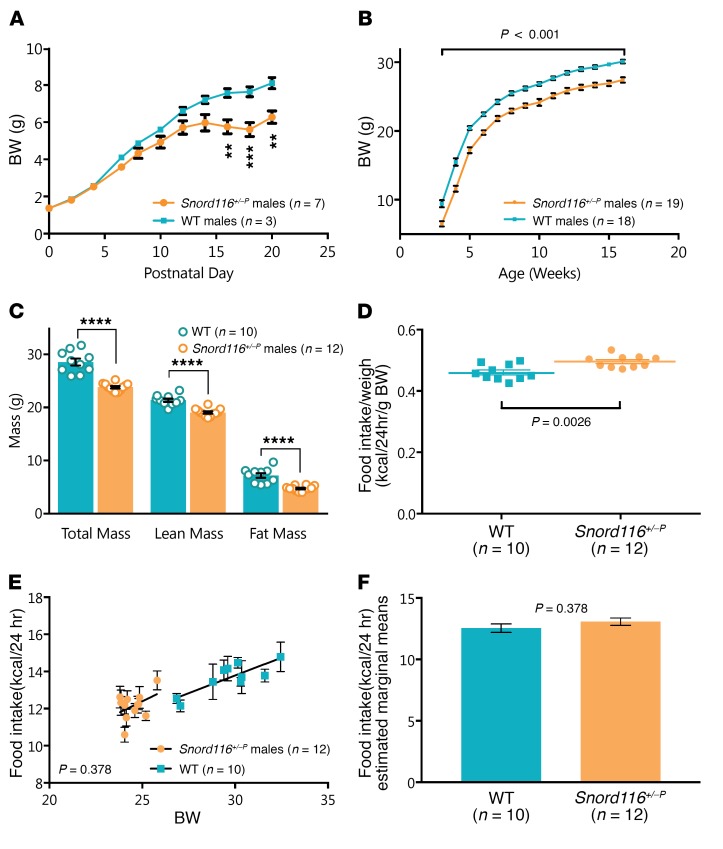
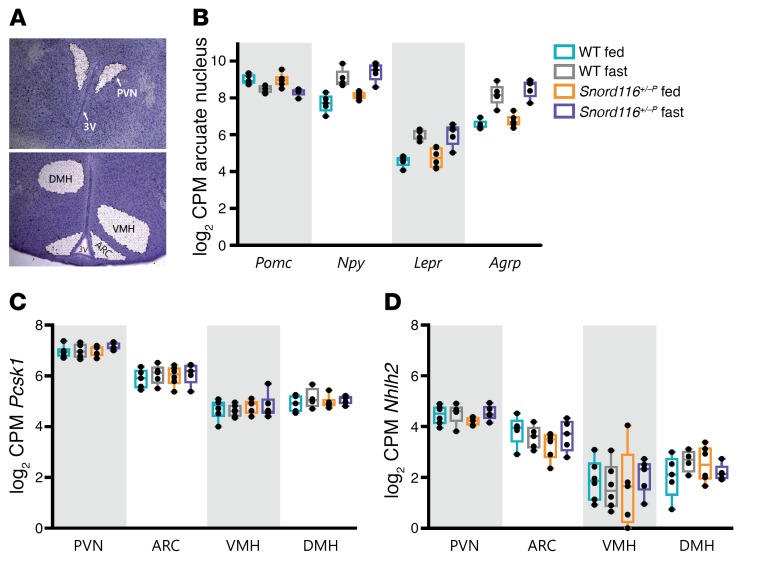
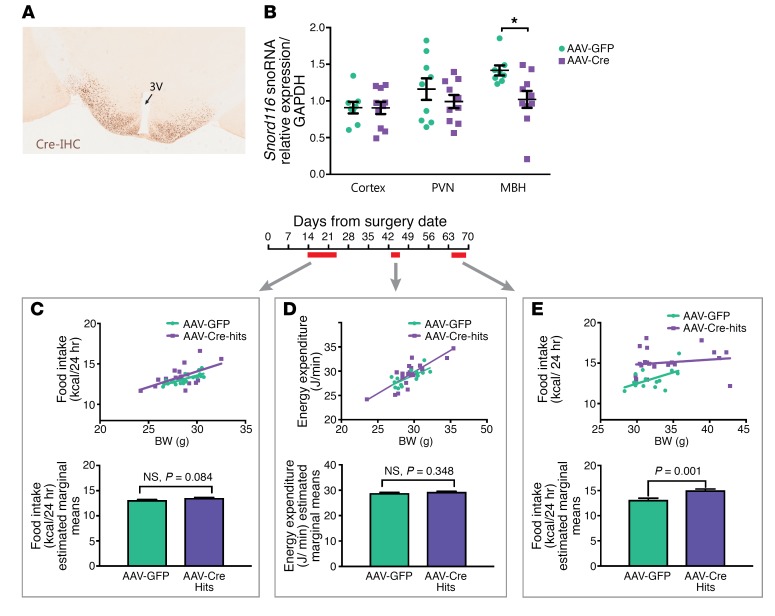
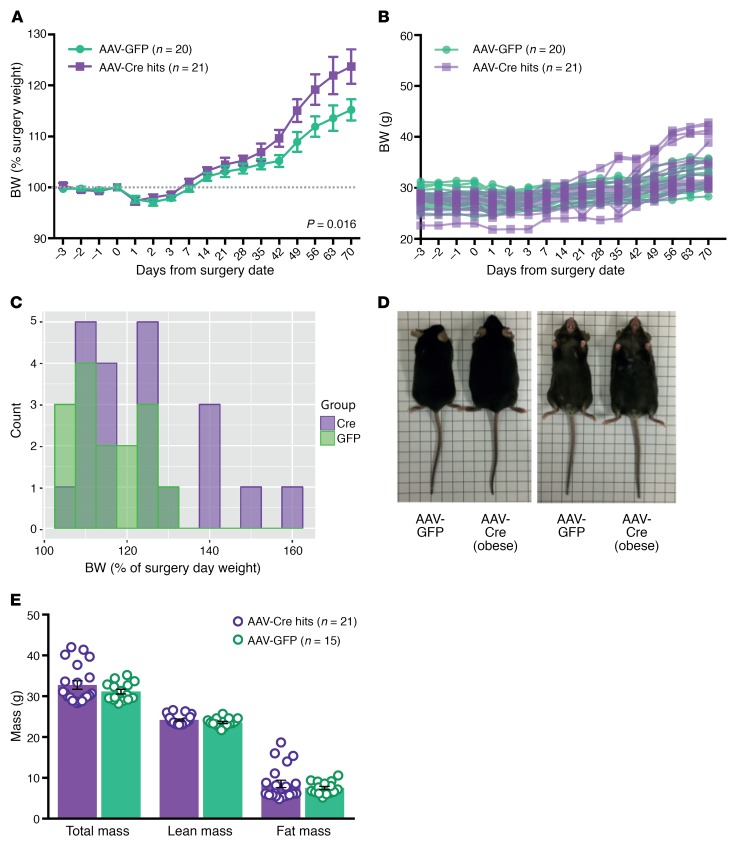
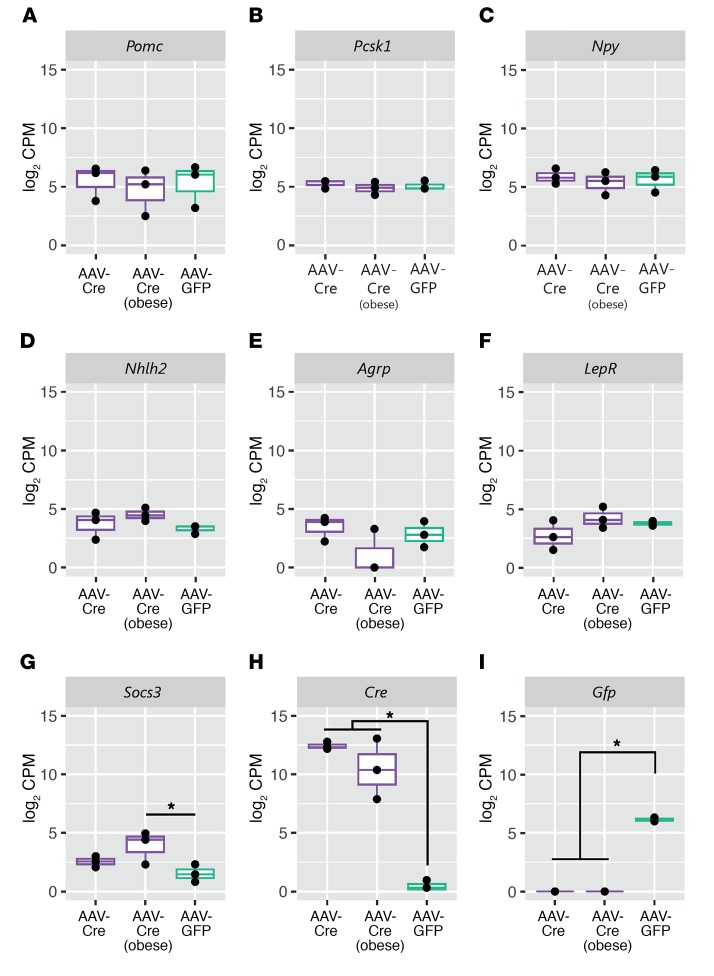
Profound hyperphagia is a major disabling feature of Prader-Willi syndrome (PWS). Characterization of the mechanisms that underlie PWS-associated hyperphagia has been slowed by the paucity of animal models with increased food intake or obesity. Mice with a microdeletion encompassing the Snord116 cluster of noncoding RNAs encoded within the Prader-Willi minimal deletion critical region have previously been reported to show growth retardation and hyperphagia. Here, consistent with previous reports, we observed growth retardation in Snord116+/-P mice with a congenital paternal Snord116 deletion. However, these mice neither displayed increased food intake nor had reduced hypothalamic expression of the proprotein convertase 1 gene PCSK1 or its upstream regulator NHLH2, which have recently been suggested to be key mediators of PWS pathogenesis. Specifically, we disrupted Snord116 expression in the mediobasal hypothalamus in Snord116fl mice via bilateral stereotaxic injections of a Cre-expressing adeno-associated virus (AAV). While the Cre-injected mice had no change in measured energy expenditure, they became hyperphagic between 9 and 10 weeks after injection, with a subset of animals developing marked obesity. In conclusion, we show that selective disruption of Snord116 expression in the mediobasal hypothalamus models the hyperphagia of PWS.
Keywords: Genetics; Metabolism; Obesity.
Conflict of interest statement
Figures
Comment in
-
Hypothalamic loss of Snord116 and Prader-Willi syndrome hyperphagia: the buck stops here?J Clin Invest. 2018 Mar 1;128(3):900-902. doi: 10.1172/JCI99725. Epub 2018 Jan 29. J Clin Invest. 2018. PMID: 29376891 Free PMC article.
-
Genetics: Functional link to hyperphagia in PWS.Nat Rev Endocrinol. 2018 Apr;14(4):192. doi: 10.1038/nrendo.2018.19. Epub 2018 Feb 23. Nat Rev Endocrinol. 2018. PMID: 29472710 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
