

Prediction of enzymatic pathways by integrative pathway mapping
- PMID: 29377793
- PMCID: PMC5788505
- DOI: 10.7554/eLife.31097
Prediction of enzymatic pathways by integrative pathway mapping
Abstract
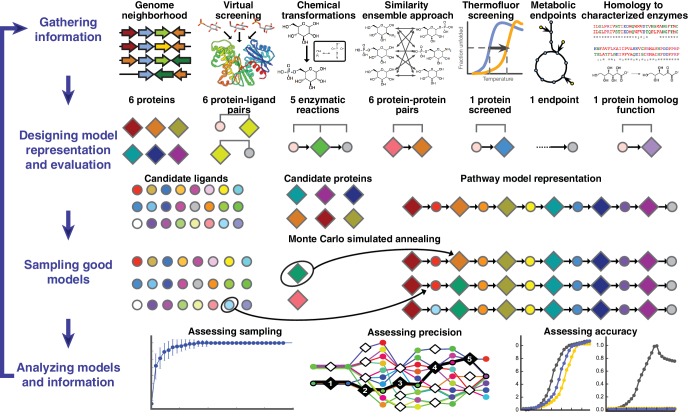
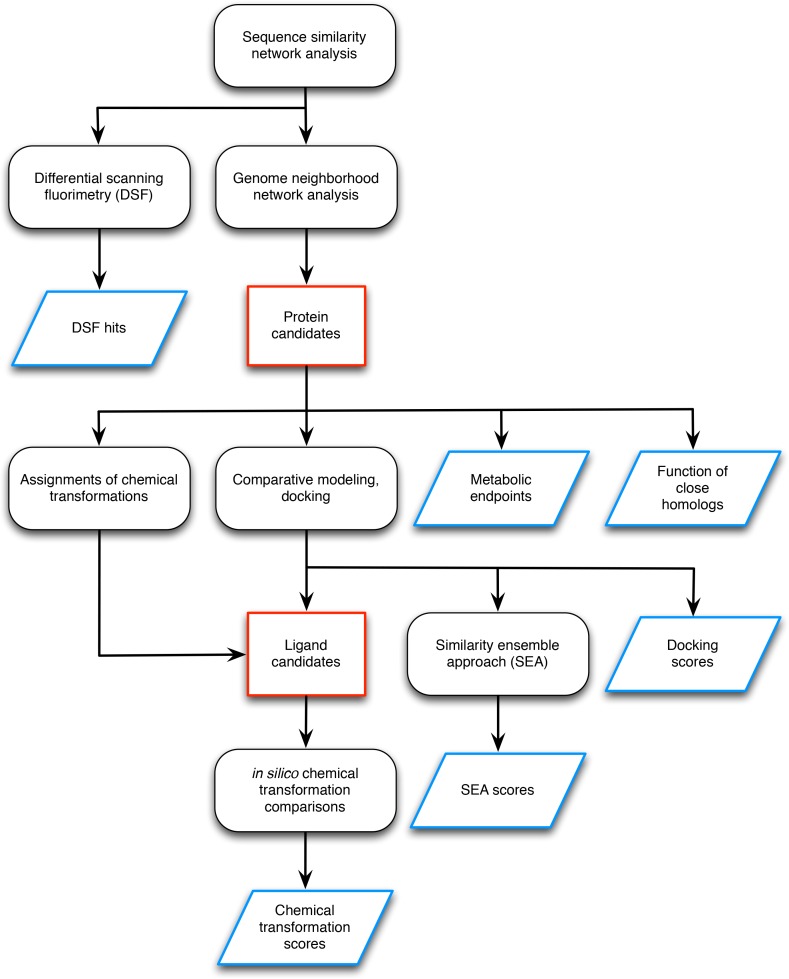
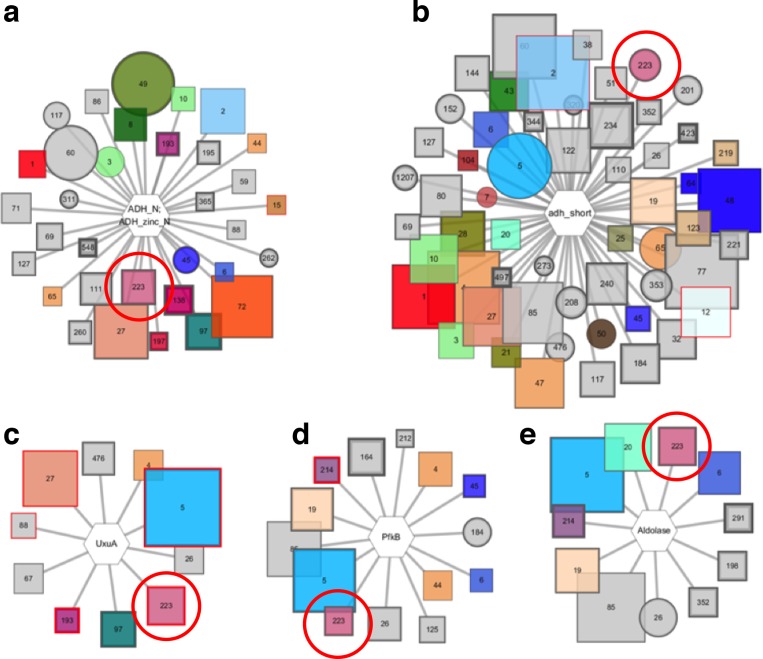
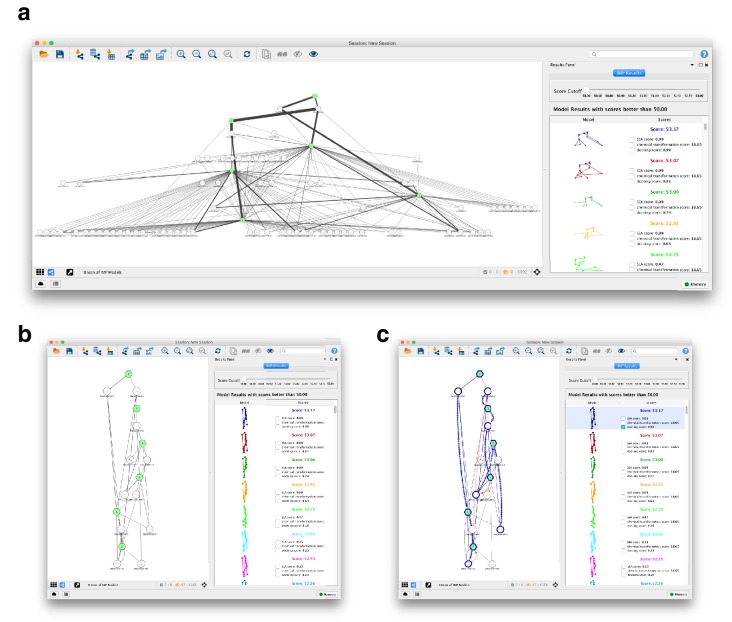
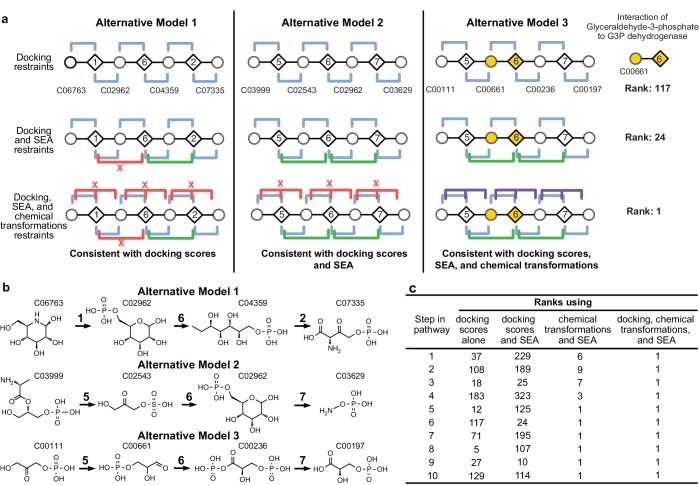
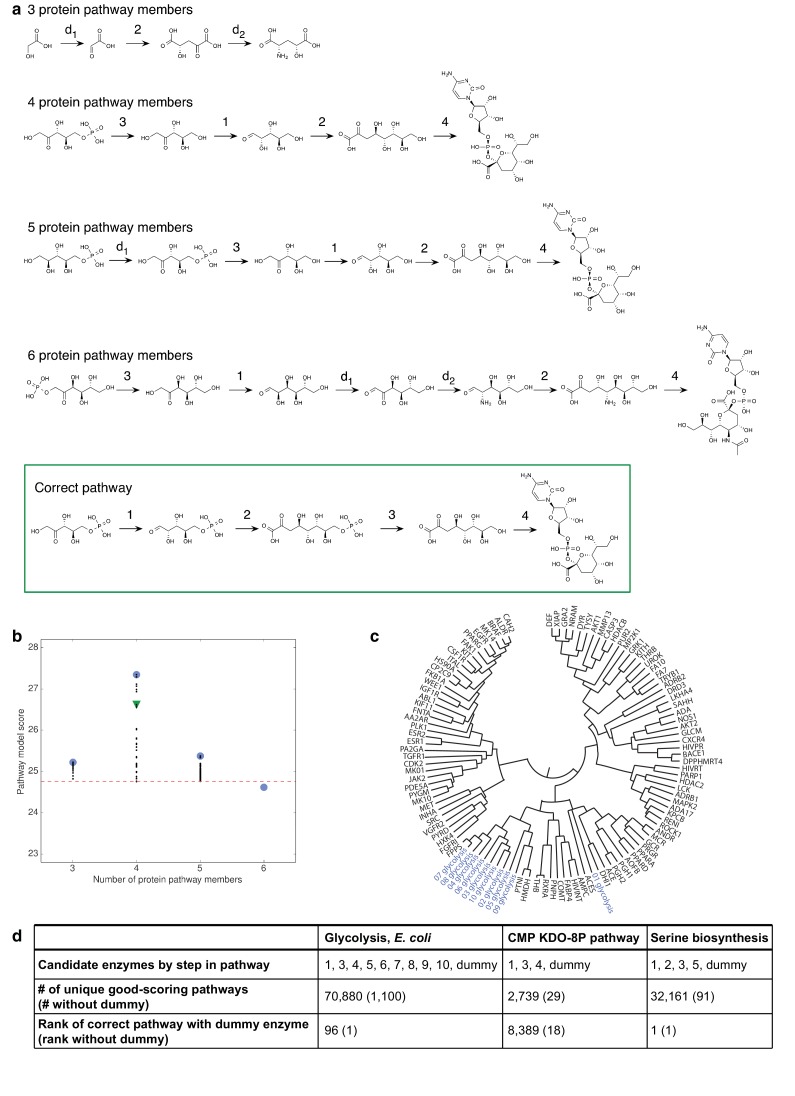
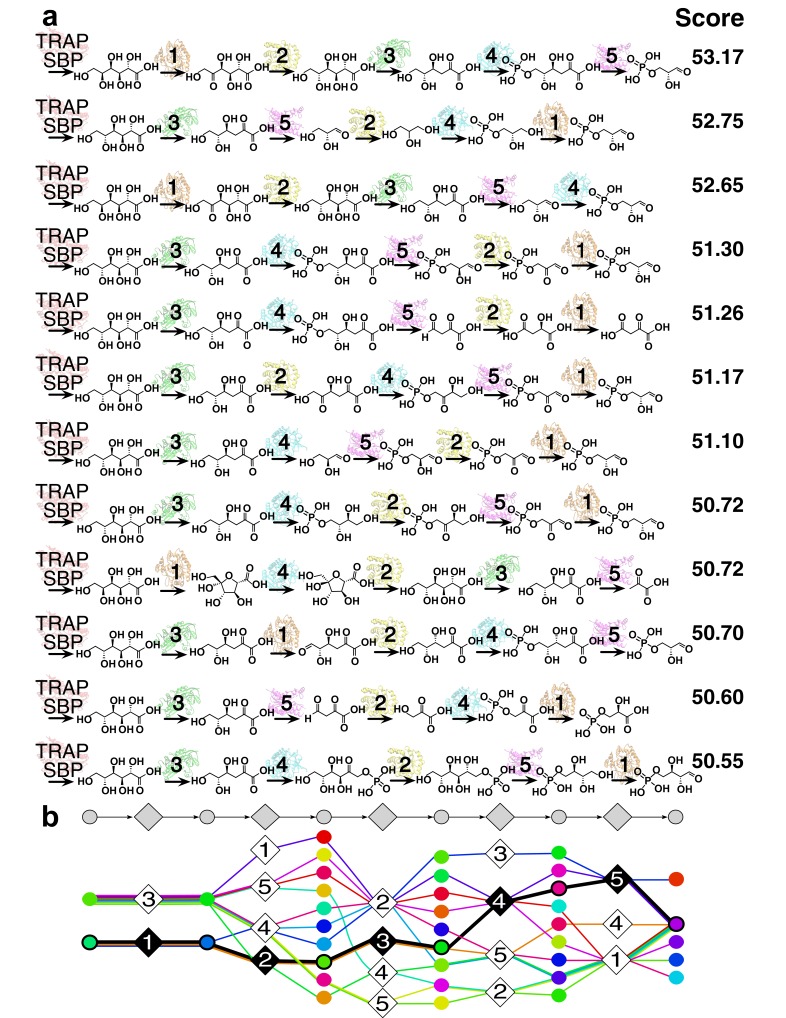
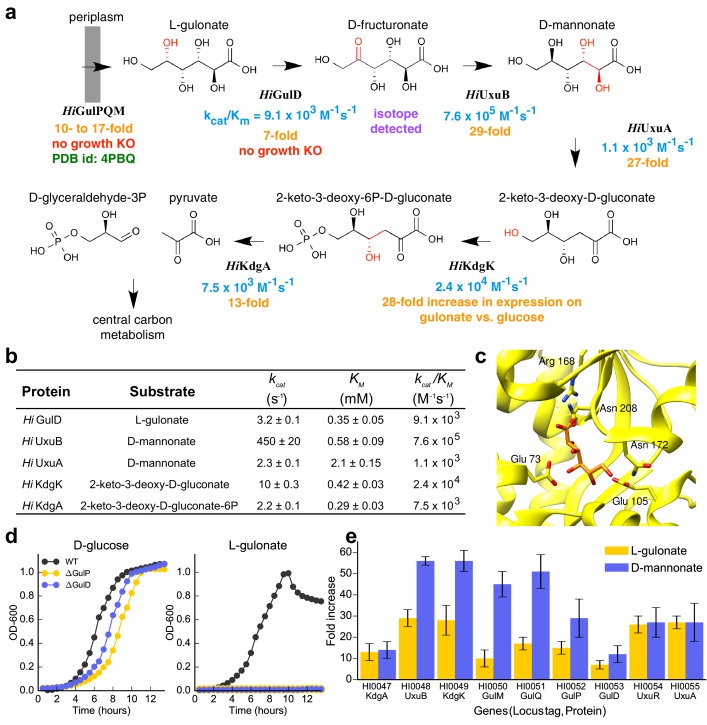
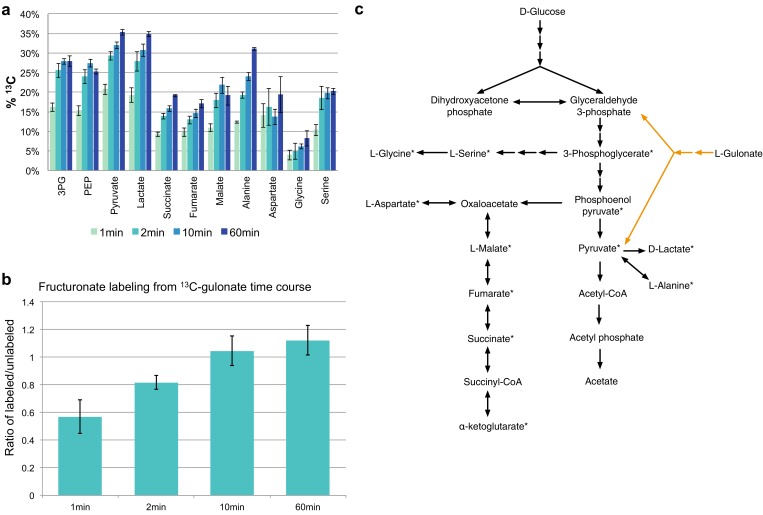
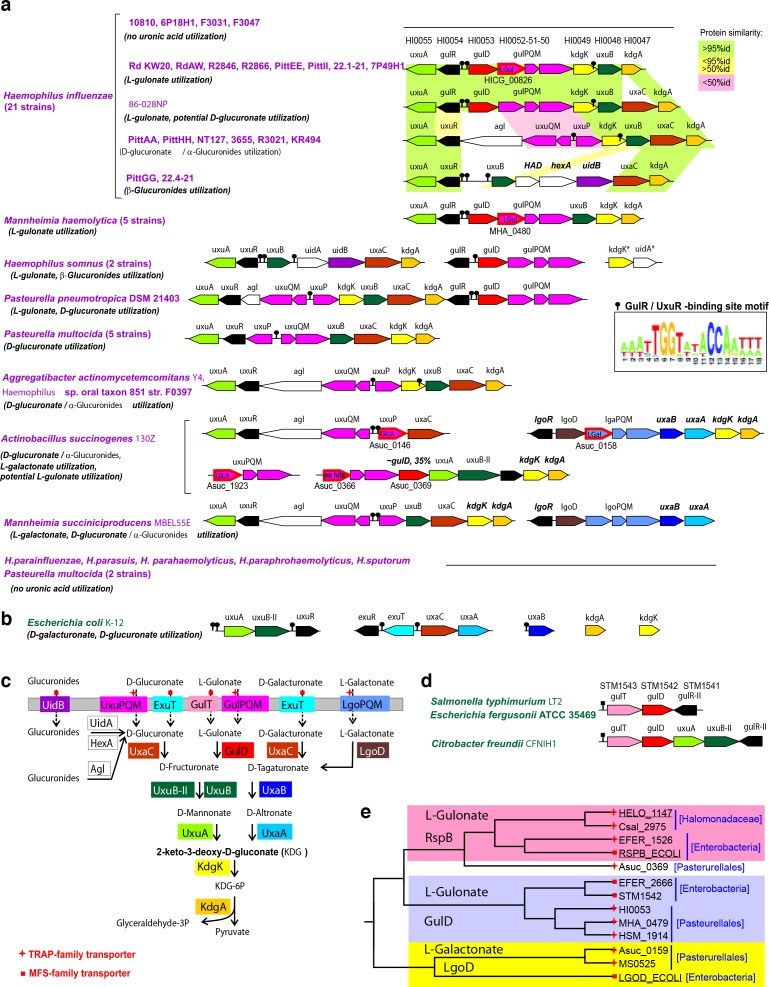
The functions of most proteins are yet to be determined. The function of an enzyme is often defined by its interacting partners, including its substrate and product, and its role in larger metabolic networks. Here, we describe a computational method that predicts the functions of orphan enzymes by organizing them into a linear metabolic pathway. Given candidate enzyme and metabolite pathway members, this aim is achieved by finding those pathways that satisfy structural and network restraints implied by varied input information, including that from virtual screening, chemoinformatics, genomic context analysis, and ligand -binding experiments. We demonstrate this integrative pathway mapping method by predicting the L-gulonate catabolic pathway in Haemophilus influenzae Rd KW20. The prediction was subsequently validated experimentally by enzymology, crystallography, and metabolomics. Integrative pathway mapping by satisfaction of structural and network restraints is extensible to molecular networks in general and thus formally bridges the gap between structural biology and systems biology.
Keywords: biophysics; computational biology; enzyme function annotation; integrative pathway mapping; l-gulonate catabolic pathway; none; pathway prediction; structural biology; structure based pathway discovery; systems biology.
© 2017, Calhoun et al.
Conflict of interest statement
SC, MK, DW, BS, SZ, DR, MV, NA, HL, MO, DS, JM, DR, SA, AO, JG, BS, AS No competing interests declared, MJ Consultant to and stockholder of Schrodinger LLC, which licenses, develops, and distributes some of the software used in this work
Figures

References
-
- Besnard J, Ruda GF, Setola V, Abecassis K, Rodriguiz RM, Huang XP, Norval S, Sassano MF, Shin AI, Webster LA, Simeons FR, Stojanovski L, Prat A, Seidah NG, Constam DB, Bickerton GR, Read KD, Wetsel WC, Gilbert IH, Roth BL, Hopkins AL. Automated design of ligands to polypharmacological profiles. Nature. 2012;492:215–220. doi: 10.1038/nature11691. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
