

Assessing runs of Homozygosity: a comparison of SNP Array and whole genome sequence low coverage data
- PMID: 29378520
- PMCID: PMC5789638
- DOI: 10.1186/s12864-018-4489-0
Assessing runs of Homozygosity: a comparison of SNP Array and whole genome sequence low coverage data
Abstract
Background: Runs of Homozygosity (ROH) are genomic regions where identical haplotypes are inherited from each parent. Since their first detection due to technological advances in the late 1990s, ROHs have been shedding light on human population history and deciphering the genetic basis of monogenic and complex traits and diseases. ROH studies have predominantly exploited SNP array data, but are gradually moving to whole genome sequence (WGS) data as it becomes available. WGS data, covering more genetic variability, can add value to ROH studies, but require additional considerations during analysis.
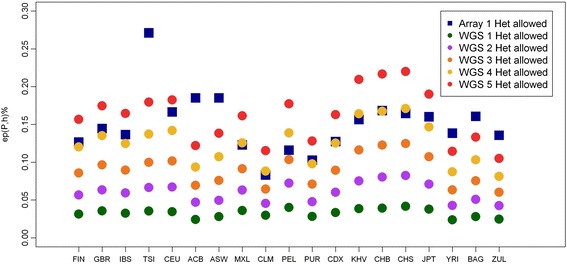
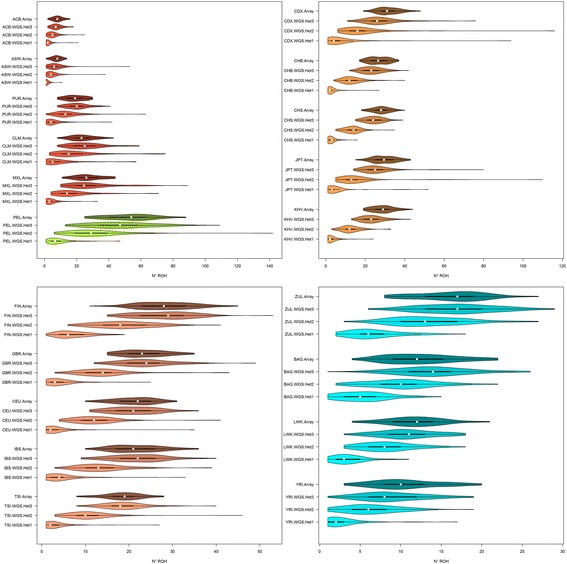
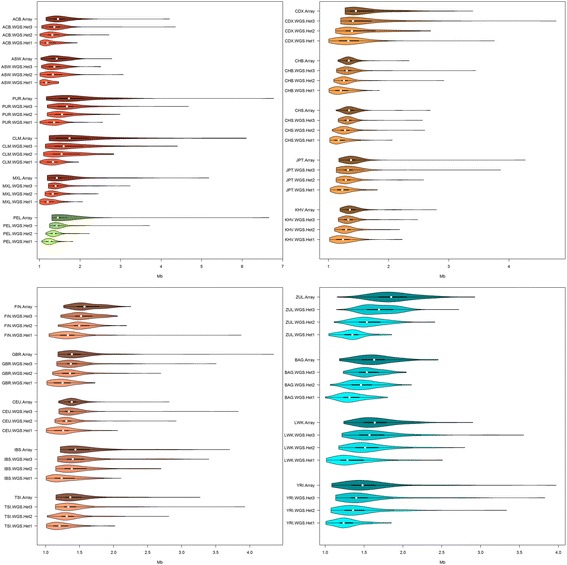
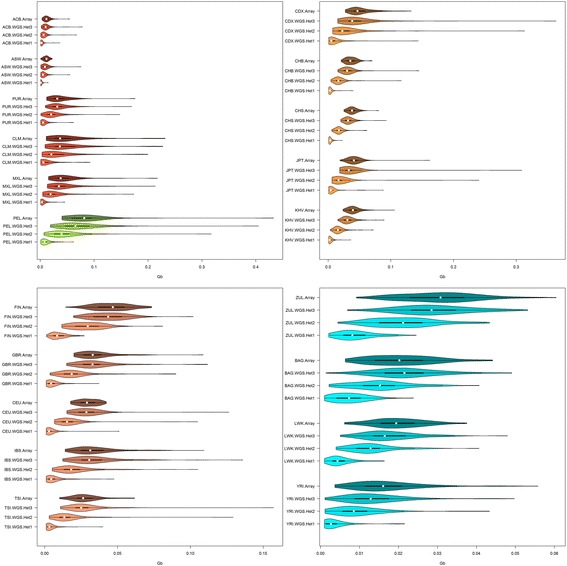
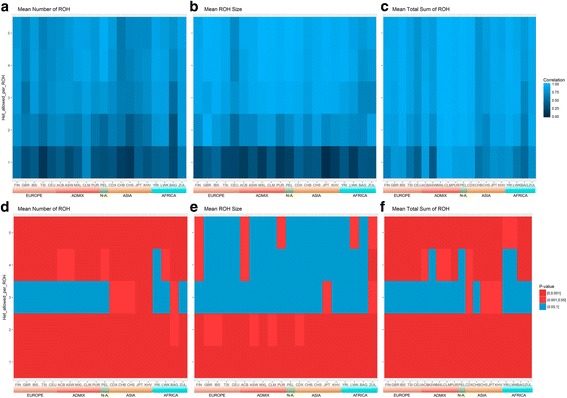
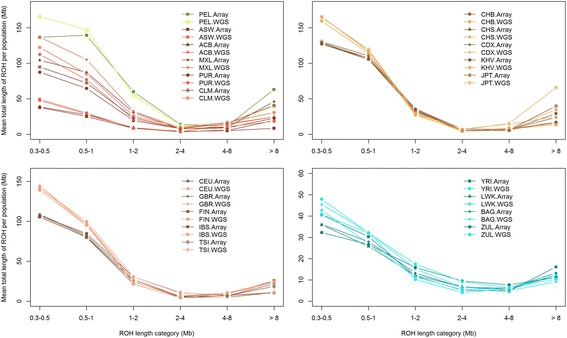
Results: Using SNP array and low coverage WGS data from 1885 individuals from 20 world populations, our aims were to compare ROH from the two datasets and to establish software conditions to get comparable results, thus providing guidelines for combining disparate datasets in joint ROH analyses. By allowing heterozygous SNPs per window, using the PLINK homozygosity function and non-parametric analysis, we were able to obtain non-significant differences in number ROH, mean ROH size and total sum of ROH between data sets using the different technologies for almost all populations.
Conclusions: By allowing 3 heterozygous SNPs per ROH when dealing with WGS low coverage data, it is possible to establish meaningful comparisons between data using SNP array and WGS low coverage technologies.
Keywords: ROH; Runs of Homozygosity; SNP array data; WGS low coverage data.
Conflict of interest statement
Ethics approval
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests
Figures
References
-
- Biraben J-N. An essay concerning mankind's demographic evolution. J Hum Evol. 1980;9:655–663. doi: 10.1016/0047-2484(80)90099-8. - DOI
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
