

Host genetic variation and its microbiome interactions within the Human Microbiome Project
- PMID: 29378630
- PMCID: PMC5789541
- DOI: 10.1186/s13073-018-0515-8
Host genetic variation and its microbiome interactions within the Human Microbiome Project
Abstract
Background: Despite the increasing recognition that microbial communities within the human body are linked to health, we have an incomplete understanding of the environmental and molecular interactions that shape the composition of these communities. Although host genetic factors play a role in these interactions, these factors have remained relatively unexplored given the requirement for large population-based cohorts in which both genotyping and microbiome characterization have been performed.
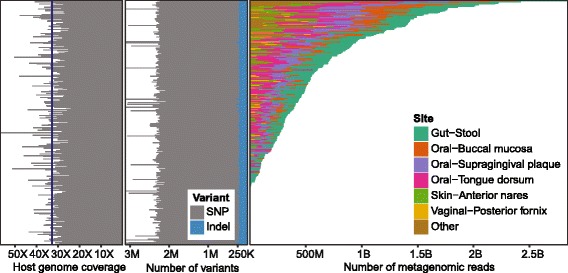
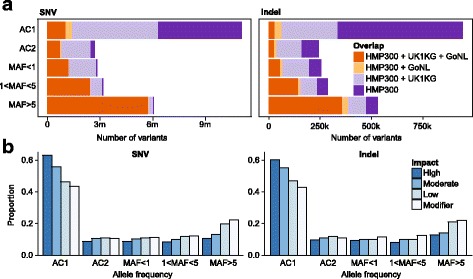
Methods: We performed whole-genome sequencing of 298 donors from the Human Microbiome Project (HMP) healthy cohort study to accompany existing deep characterization of their microbiomes at various body sites. This analysis yielded an average sequencing depth of 32x, with which we identified 27 million (M) single nucleotide variants and 2.3 M insertions-deletions.
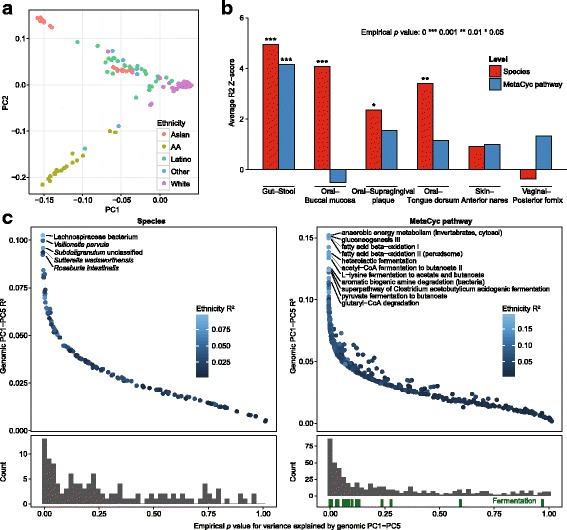
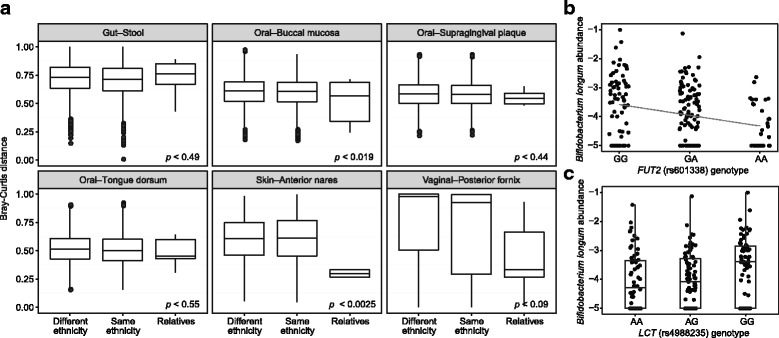
Results: Taxonomic composition and functional potential of the microbiome covaried significantly with genetic principal components in the gastrointestinal tract and oral communities, but not in the nares or vaginal microbiota. Example associations included validation of known associations between FUT2 secretor status, as well as a variant conferring hypolactasia near the LCT gene, with Bifidobacterium longum abundance in stool. The associations of microbial features with both high-level genetic attributes and single variants were specific to particular body sites, highlighting the opportunity to find unique genetic mechanisms controlling microbiome properties in the microbial communities from multiple body sites.
Conclusions: This study adds deep sequencing of host genomes to the body-wide microbiome sequences already extant from the HMP healthy cohort, creating a unique, versatile, and well-controlled reference for future studies seeking to identify host genetic modulators of the microbiome.
Keywords: Association studies; Human Microbiome Project; Human genome sequence; Microbiome and human genetics; Microbiome metagenome sequence.
Conflict of interest statement
Ethics approval and consent to participate
Recruitment protocols were approved by the appropriate institutional review boards (IRBs) at each HMP clinical site (Baylor College of Medicine, IRB protocols H-22895 (IRB 00001021) and H-22035 (IRB 00002649)); Washington University School of Medicine (IRB protocol HMP-07-001 (IRB 201105198)); and St. Louis University (IRB 15778). Written informed consent was obtained from all study participants to participate in the study and to allow data sharing through dbGaP. All participants consented for the sequencing of their own genetic material [47]. Research on human subjects was performed in accordance with the Declaration of Helsinki. The study was also reviewed by the J. Craig Venter Institute under IRB protocol 2008–084 (IRB 00003721). The study was determined to be exempt from IRB review at the Broad Institute.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Pham TA, Clare S, Goulding D, Arasteh JM, Stares MD, Browne HP, Keane JA, Page AJ, Kumasaka N, Kane L, et al. Epithelial IL-22RA1-mediated fucosylation promotes intestinal colonization resistance to an opportunistic pathogen. Cell Host Microbe. 2014;16:504–16. doi: 10.1016/j.chom.2014.08.017. - DOI - PMC - PubMed
-
- Aujoulat F, Bouvet P, Jumas-Bilak E, Jean-Pierre H, Marchandin H. Veillonella seminalis sp. nov., a novel anaerobic Gram-stain-negative coccus from human clinical samples, and emended description of the genus Veillonella. Int J Syst Evol Microbiol. 2014;64:3526–31. doi: 10.1099/ijs.0.064451-0. - DOI - PubMed
-
- Kevans D, Turpin W, Madsen K, Meddings J, Shestopaloff K, Xu W, Moreno-Hagelsieb G, Griffiths A, Silverberg MS, Paterson A, et al. Determinants of intestinal permeability in healthy first-degree relatives of individuals with Crohn’s disease. Inflamm Bowel Dis. 2015;21:879–87. doi: 10.1097/MIB.0000000000000323. - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
