

Commensal microbiota modulate gene expression in the skin
- PMID: 29378633
- PMCID: PMC5789709
- DOI: 10.1186/s40168-018-0404-9
Commensal microbiota modulate gene expression in the skin
Abstract
Background: The skin harbors complex communities of resident microorganisms, yet little is known of their physiological roles and the molecular mechanisms that mediate cutaneous host-microbe interactions. Here, we profiled skin transcriptomes of mice reared in the presence and absence of microbiota to elucidate the range of pathways and functions modulated in the skin by the microbiota.
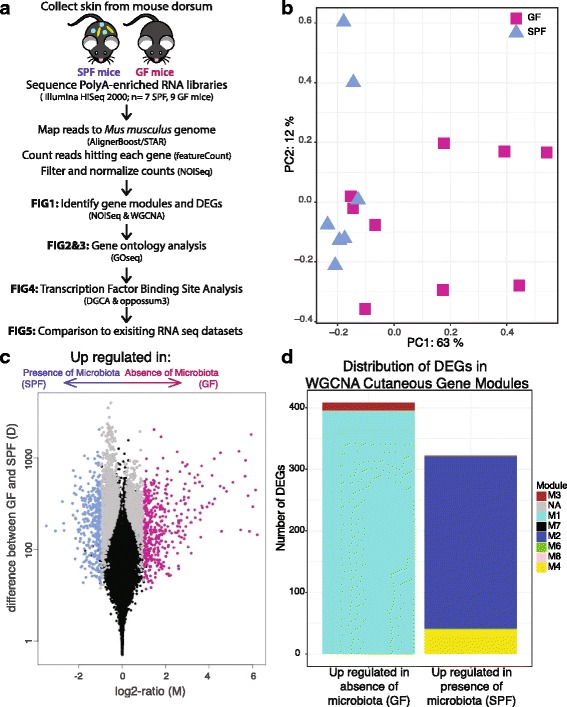
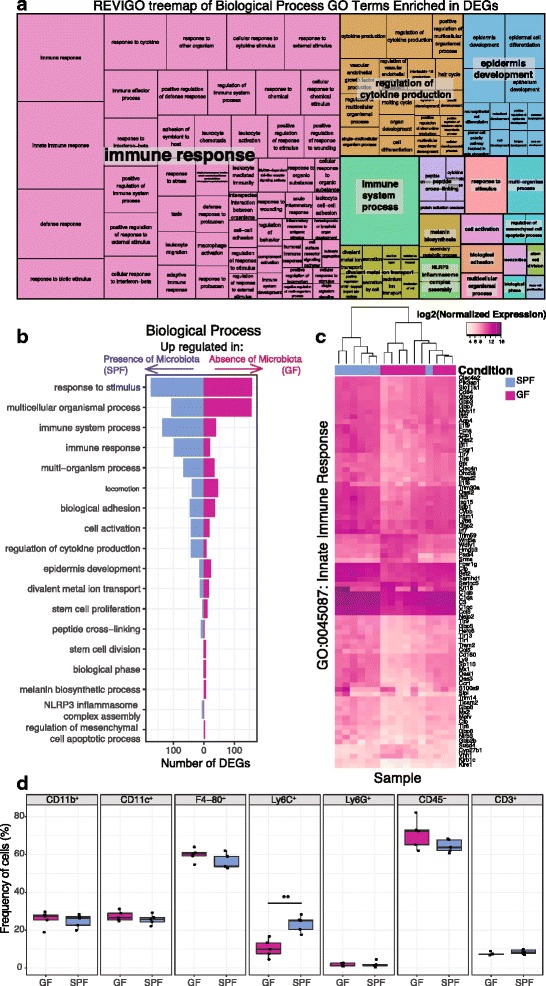
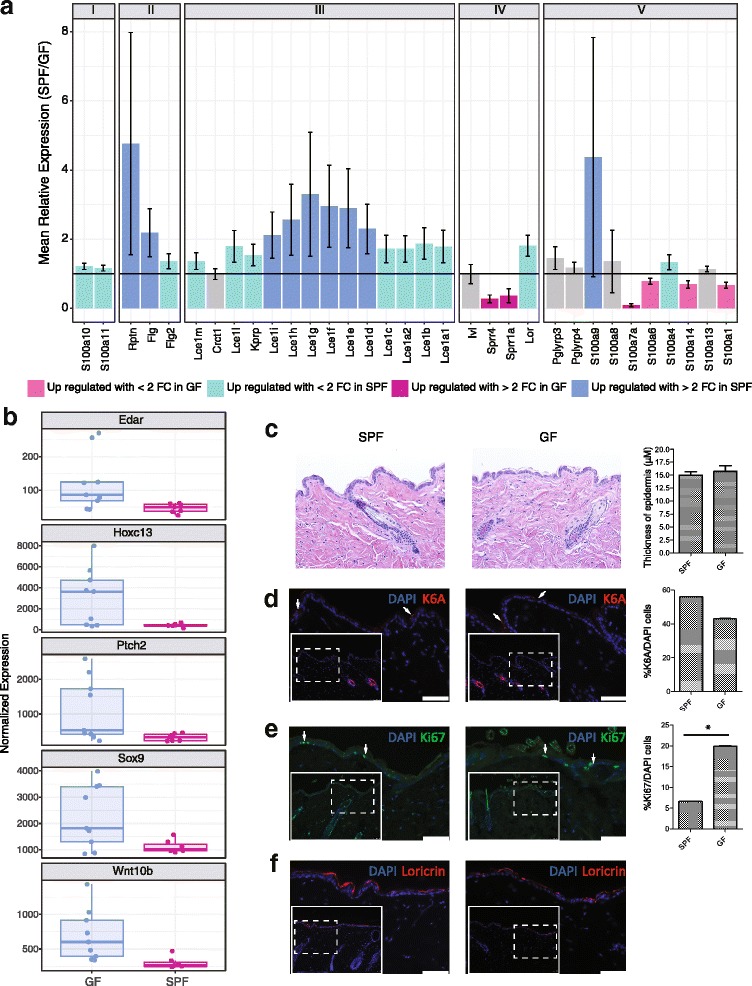
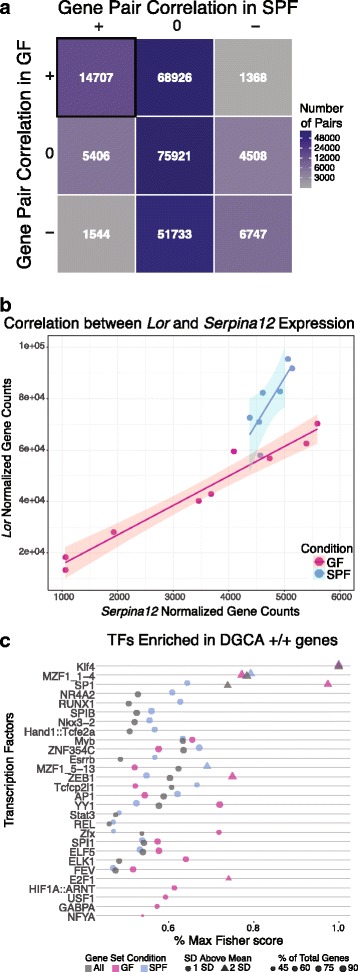
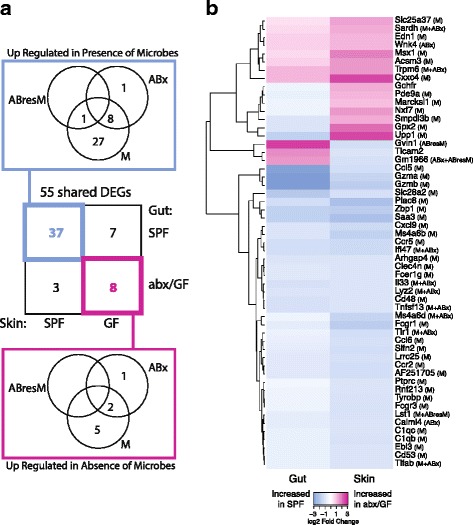
Results: A total of 2820 genes were differentially regulated in response to microbial colonization and were enriched in gene ontology (GO) terms related to the host-immune response and epidermal differentiation. Innate immune response genes and genes involved in cytokine activity were generally upregulated in response to microbiota and included genes encoding toll-like receptors, antimicrobial peptides, the complement cascade, and genes involved in IL-1 family cytokine signaling and homing of T cells. Our results also reveal a role for the microbiota in modulating epidermal differentiation and development, with differential expression of genes in the epidermal differentiation complex (EDC). Genes with correlated co-expression patterns were enriched in binding sites for the transcription factors Klf4, AP-1, and SP-1, all implicated as regulators of epidermal differentiation. Finally, we identified transcriptional signatures of microbial regulation common to both the skin and the gastrointestinal tract.
Conclusions: With this foundational approach, we establish a critical resource for understanding the genome-wide implications of microbially mediated gene expression in the skin and emphasize prospective ways in which the microbiome contributes to skin health and disease.
Keywords: Cutaneous transcriptome; Germ-free mice; Host-microbe interactions; RNA sequencing; Skin microbiome.
Conflict of interest statement
Ethics approval and consent to participate
All mouse experiments were approved by the University of Pennsylvania Institutional Animal Care and Use Committee (IACUC).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Grice EA, Kong HH, Conlan S, Deming CB, Davis J, Young AC, NISC Comparative Sequencing Program. Bouffard GG, Blakesley RW, Murray PR, Green ED, Turner ML, Segre JA. Topographical and temporal diversity of the human skin microbiome. Science. 2009;324:1190–1192. doi: 10.1126/science.1171700. - DOI - PMC - PubMed
-
- Hannigan GD, Meisel JS, Tyldsley AS, Zheng Q, Hodkinson BP, SanMiguel AJ, Minot S, Bushman FD, Grice EA. The human skin double-stranded DNA virome: topographical and temporal diversity, genetic enrichment, and dynamic associations with the host microbiome. MBio. 2015;6:e01578–e01515. doi: 10.1128/mBio.01578-15. - DOI - PMC - PubMed
-
- Lai Y, Di Nardo A, Nakatsuji T, Leichtle A, Yang Y, Cogen AL, Wu Z-R, Hooper LV, Schmidt RR, Aulock Von S, Radek KA, Huang C-M, Ryan AF, Gallo RL. Commensal bacteria regulate toll-like receptor 3-dependent inflammation after skin injury. Nat Med. 2009;15:1377–1382. doi: 10.1038/nm.2062. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
- R01 AI074878/AI/NIAID NIH HHS/United States
- K08 AR065577/AR/NIAMS NIH HHS/United States
- R01-NR015639/NR/NINR NIH HHS/United States
- R01 AR066663/AR/NIAMS NIH HHS/United States
- AI061570/NH/NIH HHS/United States
- R01 AI095466/AI/NIAID NIH HHS/United States
- AI083480/NH/NIH HHS/United States
- P30 AR069589/AR/NIAMS NIH HHS/United States
- P30-AR057217/AR/NIAMS NIH HHS/United States
- R00-AR060873/AR/NIAMS NIH HHS/United States
- R01 AI102942/AI/NIAID NIH HHS/United States
- AI095608/NH/NIH HHS/United States
- R21 AI087990/AI/NIAID NIH HHS/United States
- R01 NR015639/NR/NINR NIH HHS/United States
- T32 AR007465/AR/NIAMS NIH HHS/United States
- AI102942/NH/NIH HHS/United States
- R01-AR066663/AR/NIAMS NIH HHS/United States
- AI097333/NH/NIH HHS/United States
- AI087990/NH/NIH HHS/United States
- R21 AI083480/AI/NIAID NIH HHS/United States
- U01 AI095608/AI/NIAID NIH HHS/United States
- T32 HG000046/HG/NHGRI NIH HHS/United States
- R01 AI061570/AI/NIAID NIH HHS/United States
- P30 AR057217/AR/NIAMS NIH HHS/United States
- AI074878/NH/NIH HHS/United States
- R01 AI097333/AI/NIAID NIH HHS/United States
- R00 AR060873/AR/NIAMS NIH HHS/United States
- AI095466/NH/NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
