

Widespread transcriptional pausing and elongation control at enhancers
- PMID: 29378787
- PMCID: PMC5828392
- DOI: 10.1101/gad.309351.117
Widespread transcriptional pausing and elongation control at enhancers
Abstract
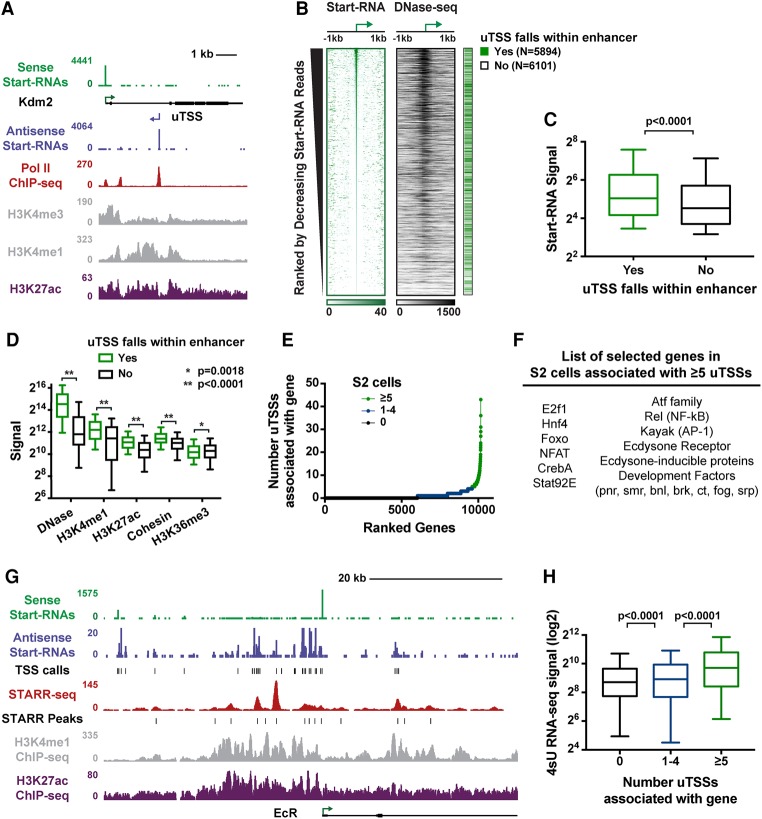
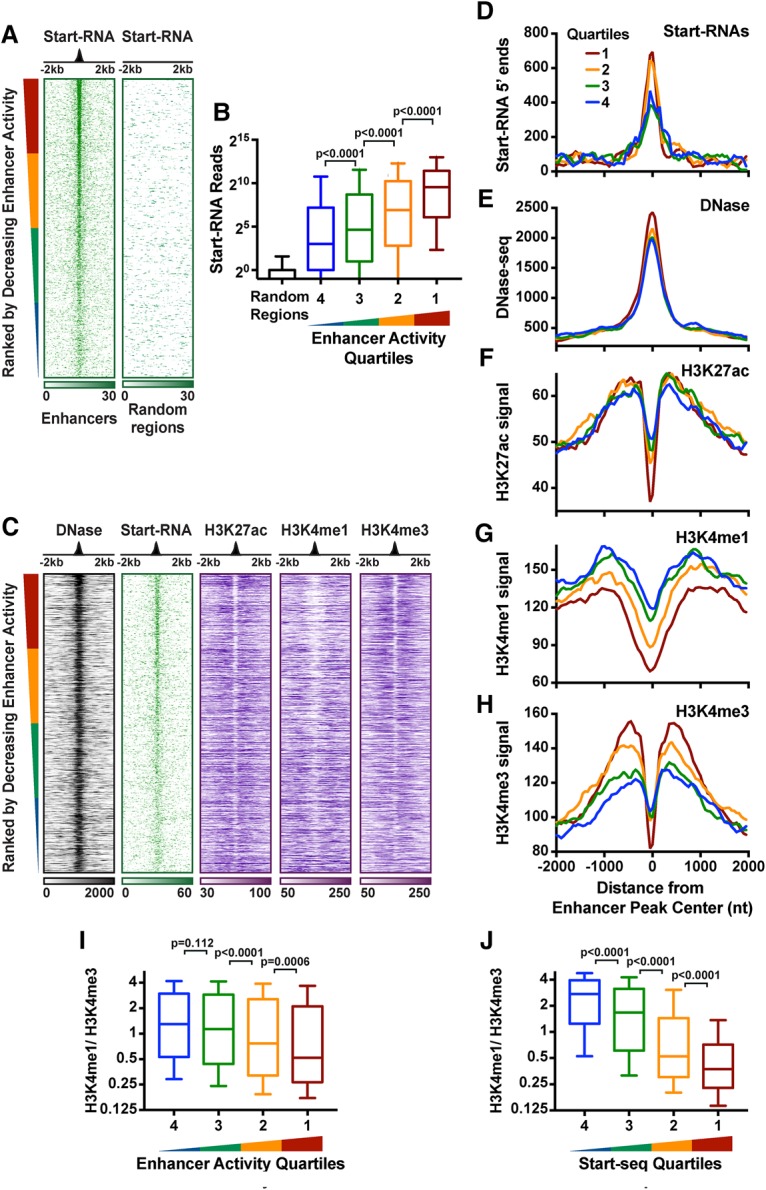
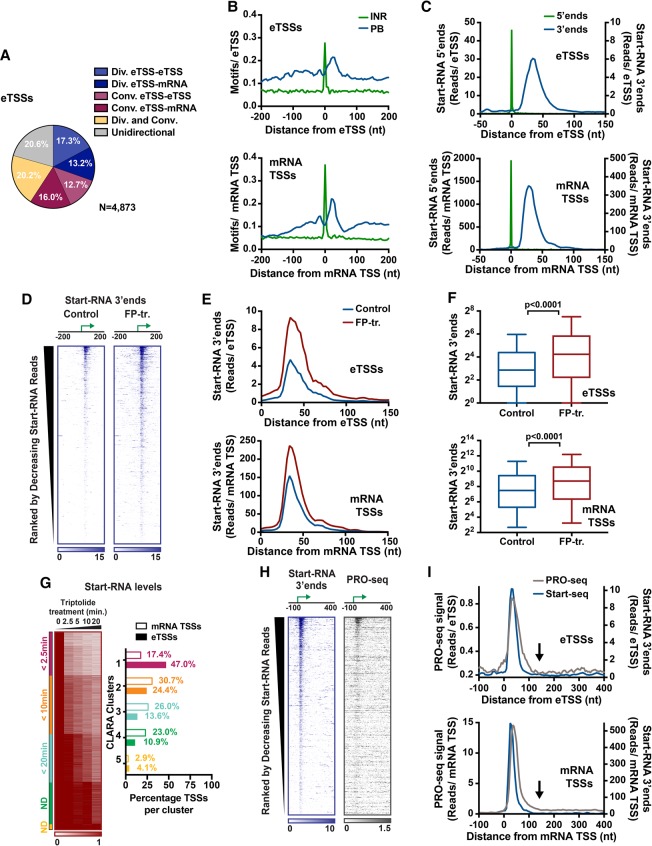
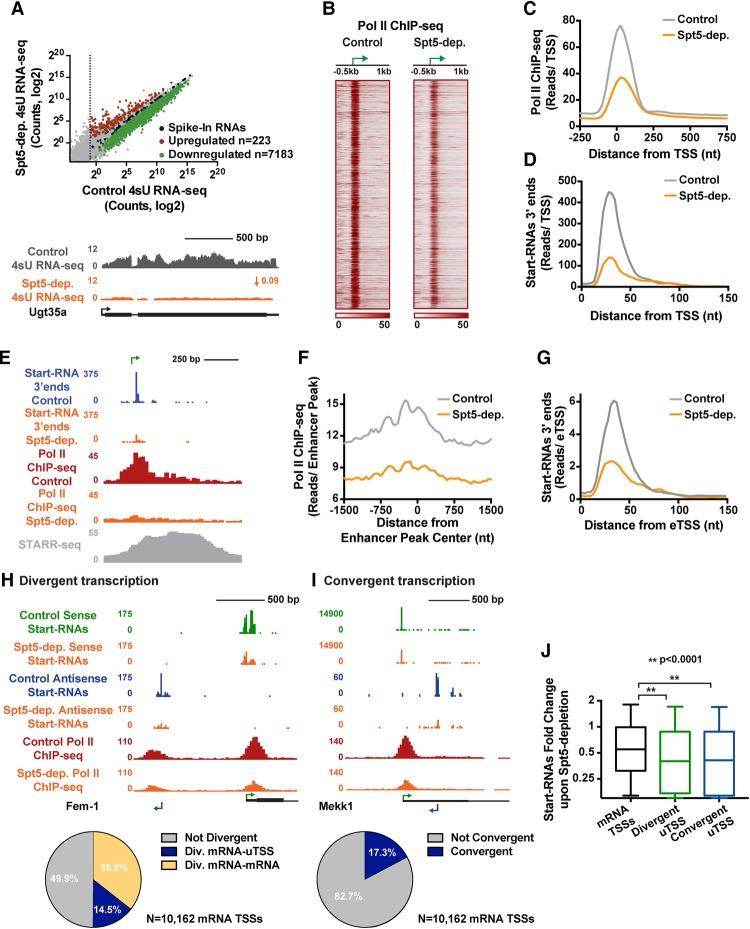
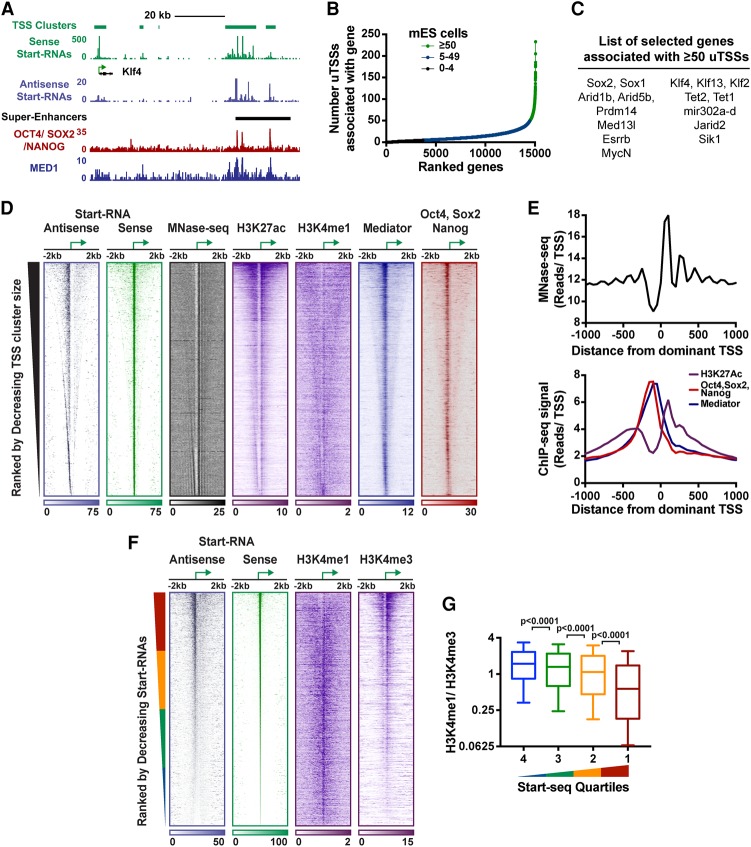
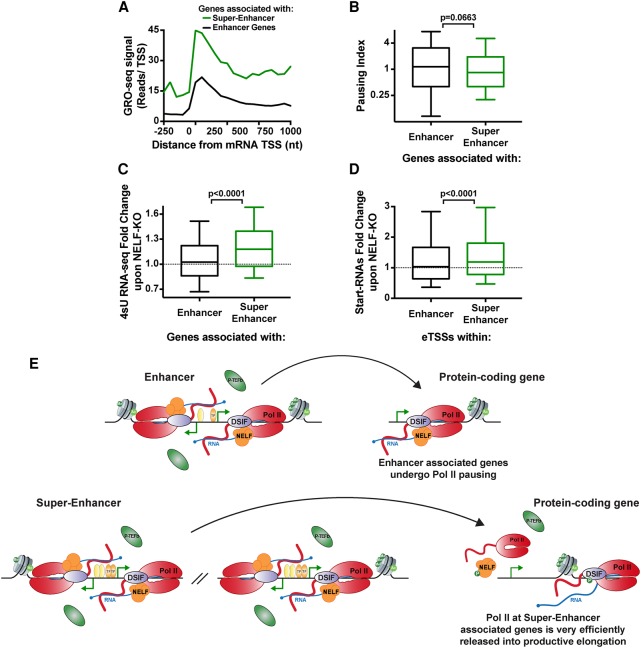
Regulation by gene-distal enhancers is critical for cell type-specific and condition-specific patterns of gene expression. Thus, to understand the basis of gene activity in a given cell type or tissue, we must identify the precise locations of enhancers and functionally characterize their behaviors. Here, we demonstrate that transcription is a nearly universal feature of enhancers in Drosophila and mammalian cells and that nascent RNA sequencing strategies are optimal for identification of both enhancers and superenhancers. We dissect the mechanisms governing enhancer transcription and discover remarkable similarities to transcription at protein-coding genes. We show that RNA polymerase II (RNAPII) undergoes regulated pausing and release at enhancers. However, as compared with mRNA genes, RNAPII at enhancers is less stable and more prone to early termination. Furthermore, we found that the level of histone H3 Lys4 (H3K4) methylation at enhancers corresponds to transcriptional activity such that highly active enhancers display H3K4 trimethylation rather than the H3K4 monomethylation considered a hallmark of enhancers. Finally, our work provides insights into the unique characteristics of superenhancers, which stimulate high-level gene expression through rapid pause release; interestingly, this property renders associated genes resistant to the loss of factors that stabilize paused RNAPII.
Keywords: P-TEFb; Pol II pausing; enhancers; superenhancers; termination; transcription.
© 2018 Henriques et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
Comment in
-
Enhancer transcription: what, where, when, and why?Genes Dev. 2018 Jan 1;32(1):1-3. doi: 10.1101/gad.311605.118. Genes Dev. 2018. PMID: 29440223 Free PMC article.
-
Gene expression: Developmental enhancers in action.Nat Rev Mol Cell Biol. 2018 Apr;19(4):210. doi: 10.1038/nrm.2018.15. Epub 2018 Feb 21. Nat Rev Mol Cell Biol. 2018. PMID: 29463930 No abstract available.
-
Gene expression: Developmental enhancers in action.Nat Rev Genet. 2018 Apr;19(4):187. doi: 10.1038/nrg.2018.13. Epub 2018 Feb 26. Nat Rev Genet. 2018. PMID: 29479083 No abstract available.
References
-
- Arnold CD, Gerlach D, Stelzer C, Boryń ŁM, Rath M, Stark A. 2013. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science 339: 1074–1077. - PubMed
-
- Austenaa LMI, Barozzi I, Simonatto M, Masella S, Chiara Della G, Ghisletti S, Curina A, de Wit E, Bouwman BAM, de Pretis S, et al. 2015. Transcription of mammalian cis-regulatory elements is restrained by actively enforced early termination. Mol Cell 60: 460–474. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases