

Aryl hydrocarbon receptor signaling modulates antiviral immune responses: ligand metabolism rather than chemical source is the stronger predictor of outcome
- PMID: 29379138
- PMCID: PMC5789012
- DOI: 10.1038/s41598-018-20197-4
Aryl hydrocarbon receptor signaling modulates antiviral immune responses: ligand metabolism rather than chemical source is the stronger predictor of outcome
Abstract
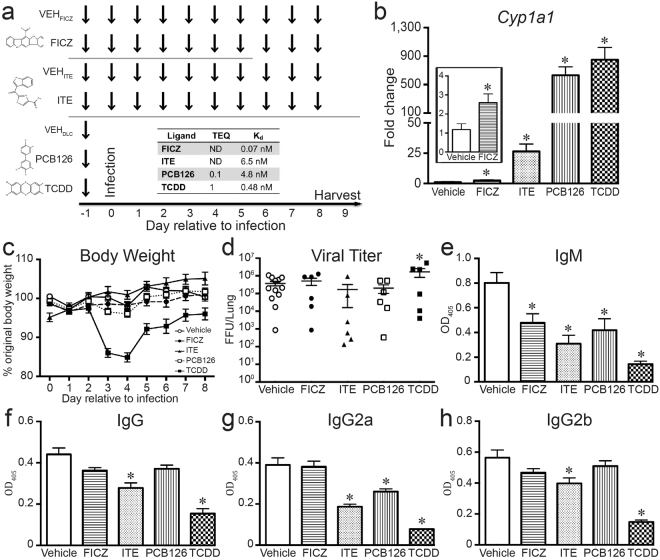
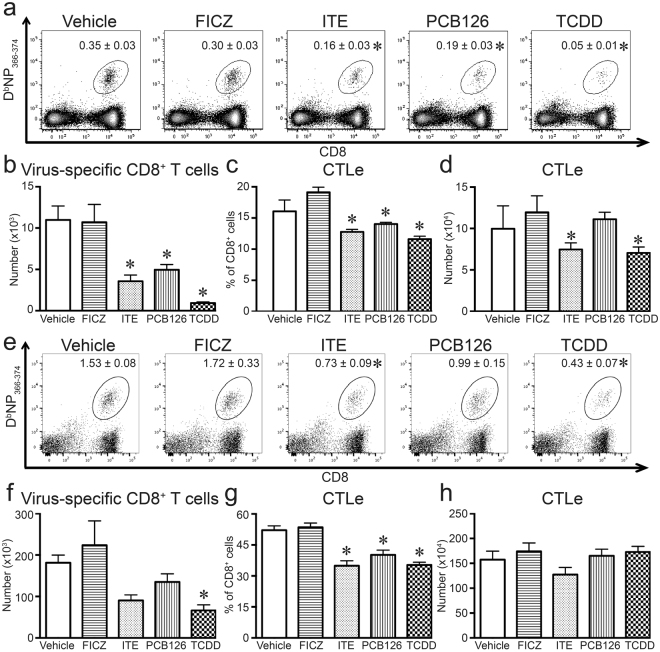
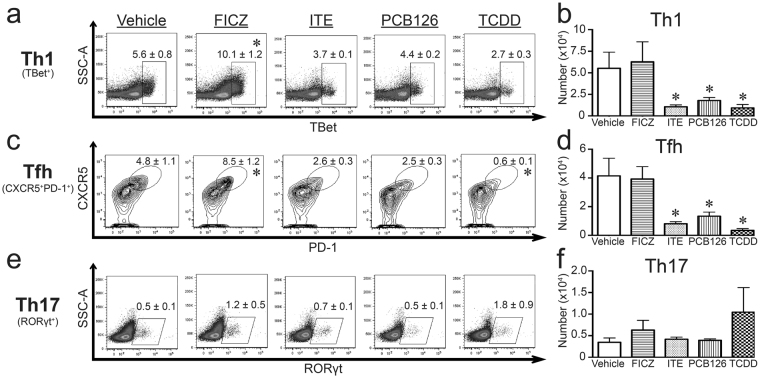
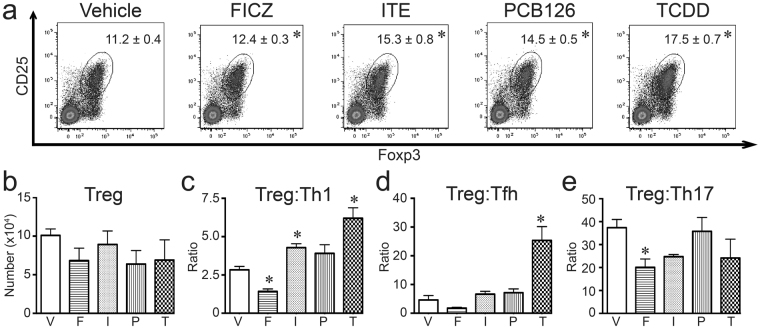
The aryl hydrocarbon receptor (AHR) offers a compelling target to modulate the immune system. AHR agonists alter adaptive immune responses, but the consequences differ across studies. We report here the comparison of four agents representing different sources of AHR ligands in mice infected with influenza A virus (IAV): TCDD, prototype exogenous AHR agonist; PCB126, pollutant with documented human exposure; ITE, novel pharmaceutical; and FICZ, degradation product of tryptophan. All four compounds diminished virus-specific IgM levels and increased the proportion of regulatory T cells. TCDD, PCB126 and ITE, but not FICZ, reduced virus-specific IgG levels and CD8+ T cell responses. Similarly, ITE, PCB126, and TCDD reduced Th1 and Tfh cells, whereas FICZ increased their frequency. In Cyp1a1-deficient mice, all compounds, including FICZ, reduced the response to IAV. Conditional Ahr knockout mice revealed that all four compounds require AHR within hematopoietic cells. Thus, differences in the immune response to IAV likely reflect variances in quality, magnitude, and duration of AHR signaling. This indicates that binding affinity and metabolism may be stronger predictors of immune effects than a compound's source of origin, and that harnessing AHR will require finding a balance between dampening immune-mediated pathologies and maintaining sufficient host defenses against infection.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
