

Many Genes-One Disease? Genetics of Nephronophthisis (NPHP) and NPHP-Associated Disorders
- PMID: 29379777
- PMCID: PMC5770800
- DOI: 10.3389/fped.2017.00287
Many Genes-One Disease? Genetics of Nephronophthisis (NPHP) and NPHP-Associated Disorders
Abstract
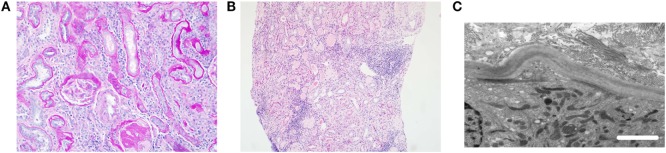
Nephronophthisis (NPHP) is a renal ciliopathy and an autosomal recessive cause of cystic kidney disease, renal fibrosis, and end-stage renal failure, affecting children and young adults. Molecular genetic studies have identified more than 20 genes underlying this disorder, whose protein products are all related to cilia, centrosome, or mitotic spindle function. In around 15% of cases, there are additional features of a ciliopathy syndrome, including retinal defects, liver fibrosis, skeletal abnormalities, and brain developmental disorders. Alongside, gene identification has arisen molecular mechanistic insights into the disease pathogenesis. The genetic causes of NPHP are discussed in terms of how they help us to define treatable disease pathways including the cyclic adenosine monophosphate pathway, the mTOR pathway, Hedgehog signaling pathways, and DNA damage response pathways. While the underlying pathology of the many types of NPHP remains similar, the defined disease mechanisms are diverse, and a personalized medicine approach for therapy in NPHP patients is likely to be required.
Keywords: DNA damage; Joubert syndrome; centrosome; cilia; ciliopathy; cyclic adenosine monophosphate; molecular genetics; nephronophthisis.
Figures
References
-
- Hildebrandt F, Strahm B, Nothwang HG, Gretz N, Schnieders B, Singh-Sawhney I, et al. Molecular genetic identification of families with juvenile nephronophthisis type 1: rate of progression to renal failure. APN Study Group. Arbeitsgemeinschaft fur Padiatrische Nephrologie. Kidney Int (1997) 51(1):261–9.10.1038/ki.1997.31 - DOI - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous