

Human Skeletal Muscle Possesses an Epigenetic Memory of Hypertrophy
- PMID: 29382913
- PMCID: PMC5789890
- DOI: 10.1038/s41598-018-20287-3
Human Skeletal Muscle Possesses an Epigenetic Memory of Hypertrophy
Abstract
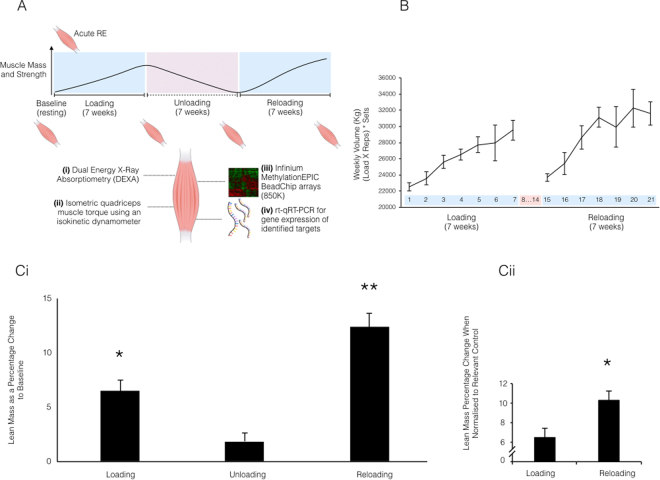
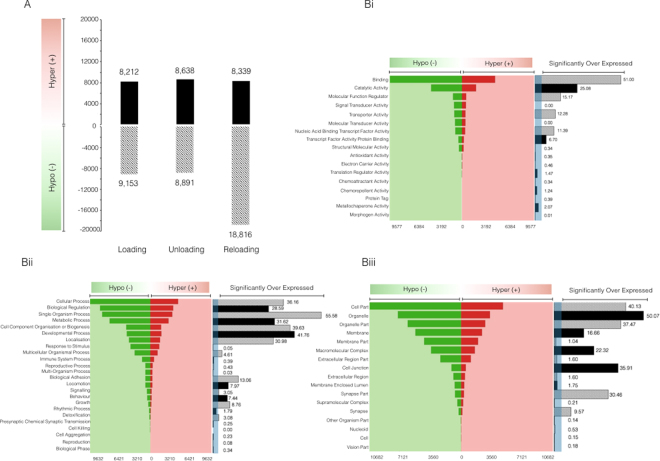
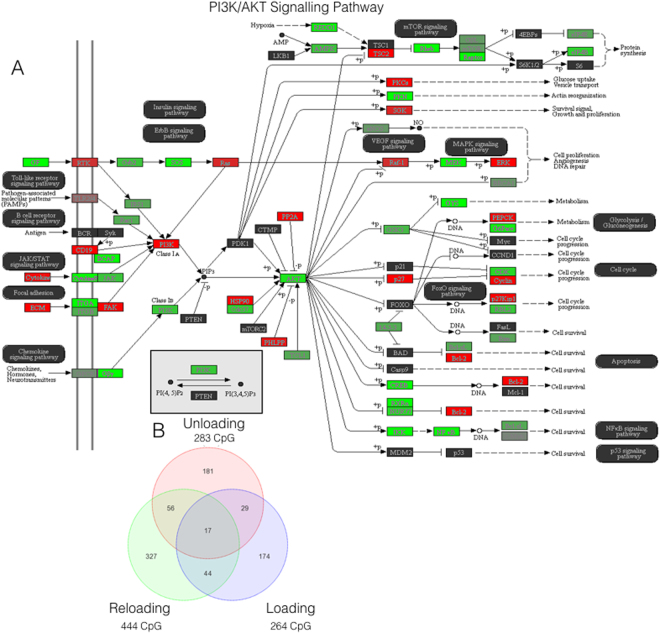
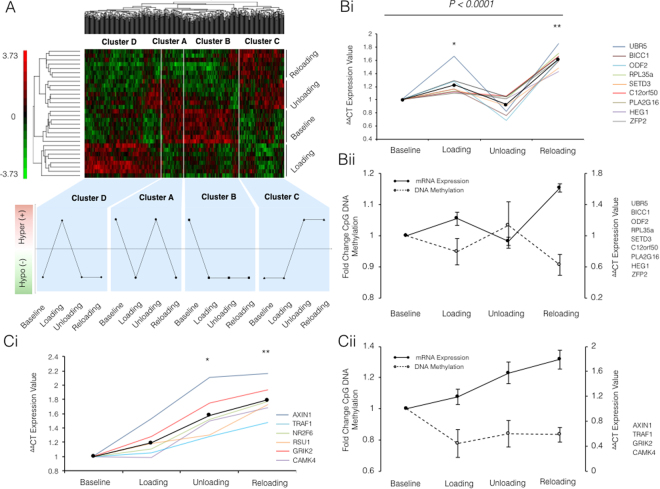
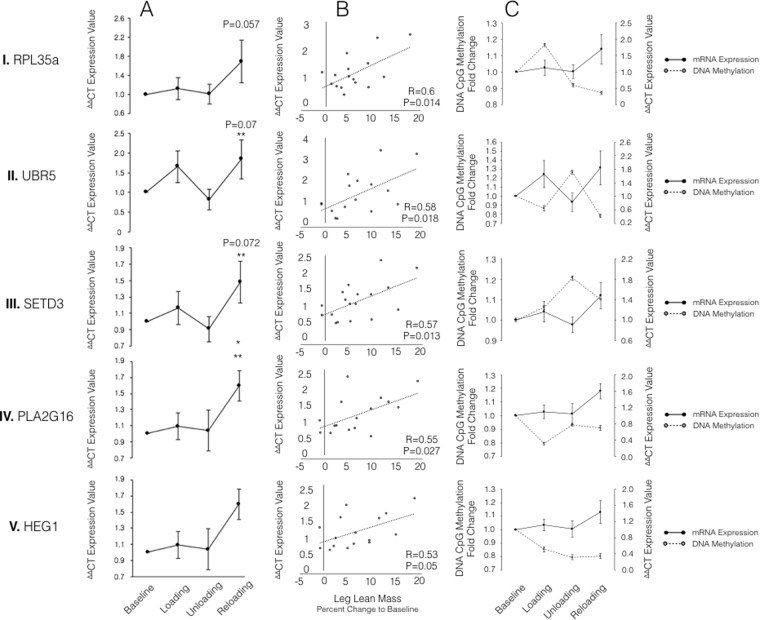
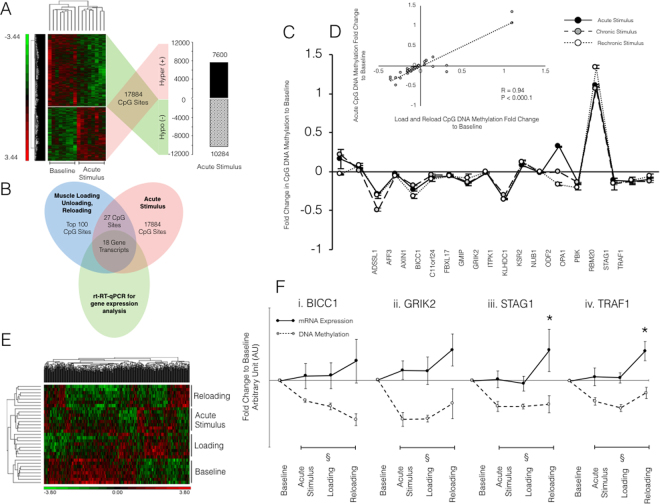
It is unknown if adult human skeletal muscle has an epigenetic memory of earlier encounters with growth. We report, for the first time in humans, genome-wide DNA methylation (850,000 CpGs) and gene expression analysis after muscle hypertrophy (loading), return of muscle mass to baseline (unloading), followed by later hypertrophy (reloading). We discovered increased frequency of hypomethylation across the genome after reloading (18,816 CpGs) versus earlier loading (9,153 CpG sites). We also identified AXIN1, GRIK2, CAMK4, TRAF1 as hypomethylated genes with enhanced expression after loading that maintained their hypomethylated status even during unloading where muscle mass returned to control levels, indicating a memory of these genes methylation signatures following earlier hypertrophy. Further, UBR5, RPL35a, HEG1, PLA2G16, SETD3 displayed hypomethylation and enhanced gene expression following loading, and demonstrated the largest increases in hypomethylation, gene expression and muscle mass after later reloading, indicating an epigenetic memory in these genes. Finally, genes; GRIK2, TRAF1, BICC1, STAG1 were epigenetically sensitive to acute exercise demonstrating hypomethylation after a single bout of resistance exercise that was maintained 22 weeks later with the largest increase in gene expression and muscle mass after reloading. Overall, we identify an important epigenetic role for a number of largely unstudied genes in muscle hypertrophy/memory.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
