

Calcium receptor signaling and citrate transport
- PMID: 29383416
- PMCID: PMC6066462
- DOI: 10.1007/s00240-018-1035-0
Calcium receptor signaling and citrate transport
Abstract
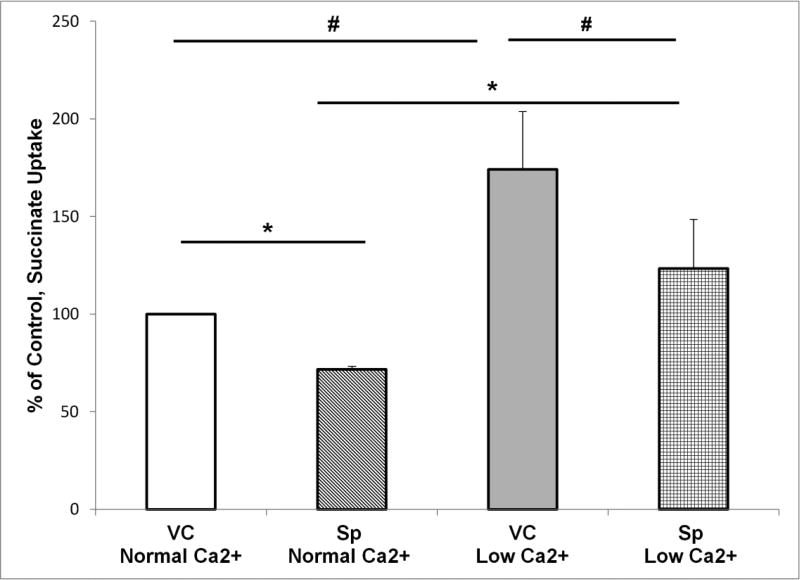
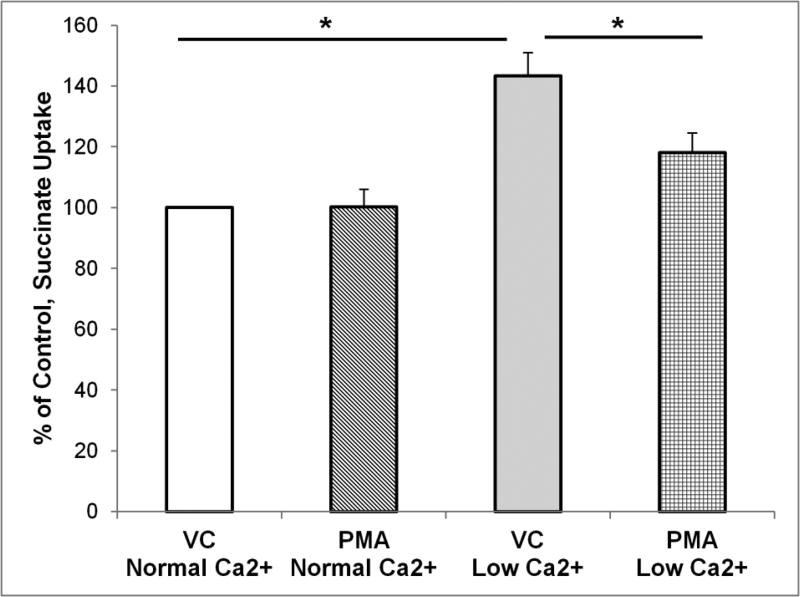
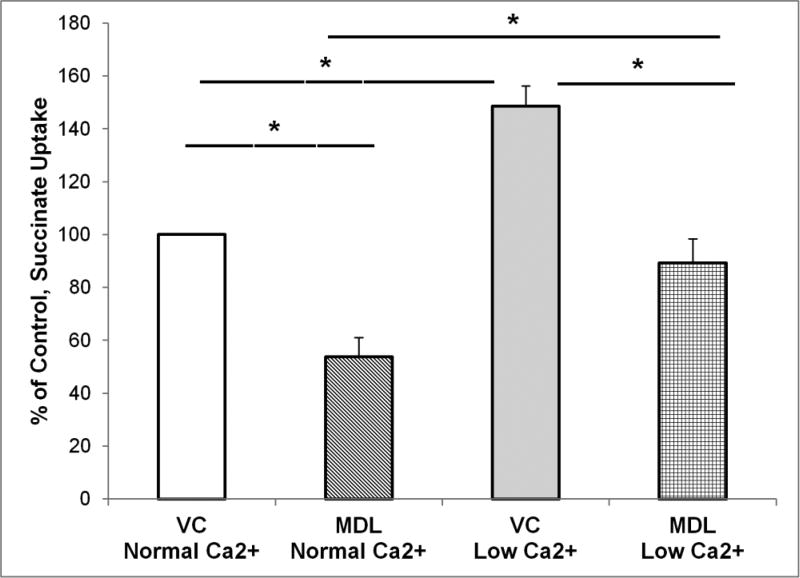
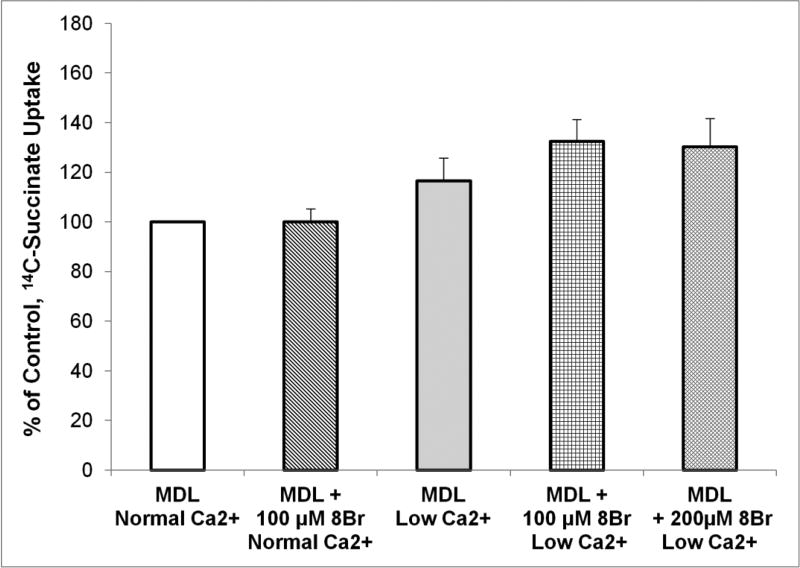
The calcium sensing receptor (CaSR) in the distal nephron decreases the propensity for calcium stones. Here we investigate if the apical CaSR in the proximal tubule also prevents stone formation acting via regulation of apical dicarboxylate and citrate transport. Urinary citrate, partially reabsorbed as a dicarboxylate in the proximal tubule lumen, inhibits stone formation by complexing calcium. We previously demonstrated a novel apical calcium-sensitive dicarboxylate transport system in OK proximal tubule cells. This calcium-sensitive process has the potential to modulate the amount of citrate available to complex increased urinary calcium. Using isotope labeled succinate uptake in OK cells along with various pharmacologic tools we examined whether the CaSR alters apical dicarboxylate transport and through which signal transduction pathways this occurs. Our results indicate that in the proximal tubule CaSR adjusts apical dicarboxylate transport, and does so via a CaSR → Gq → PKC signaling pathway. Thus, the CaSR may decrease the propensity for stone formation via actions in both proximal and distal nephron segments.
Keywords: Apical; CaSR; Calcium-sensitive; Citrate; Dicarboxylate transport; Proximal tubule.
Conflict of interest statement
Conflict of Interest: Dr Ryan Walker declares that he has no conflict of interest. Dr Shijia Zhang declares that he has no conflict of interest. Ms. Joycelynn Coleman-Barnett declares that she has no conflict of interest. Dr L. Lee Hamm declares that he has no conflict of interest. Dr Kathleen Hering-Smith declares that she has no conflict of interest.
Figures




References
-
- Quamme GA. Effect of hypercalcemia on renal tubular handling of calcium and magnesium. Can J Physiol Pharmacol. 1982;60(10):1275–1280. - PubMed
-
- Wang WH, Lu M, Hebert SC. Cytochrome P-450 metabolites mediate extracellular Ca(2+)-induced inhibition of apical K+ channels in the TAL. Am J Physiol. 1996;271(1 Pt 1):C103–111. - PubMed
-
- Topala CN, Schoeber JP, Searchfield LE, Riccardi D, Hoenderop JG, Bindels RJ. Activation of the Ca2+-sensing receptor stimulates the activity of the epithelial Ca2+ channel TRPV5. Cell Calcium. 2009;45(4):331–339. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
