

CD301b/MGL2+ Mononuclear Phagocytes Orchestrate Autoimmune Cardiac Valve Inflammation and Fibrosis
- PMID: 29386201
- PMCID: PMC5988921
- DOI: 10.1161/CIRCULATIONAHA.117.033144
CD301b/MGL2+ Mononuclear Phagocytes Orchestrate Autoimmune Cardiac Valve Inflammation and Fibrosis
Abstract
Background: Valvular heart disease is common and affects the mitral valve (MV) most frequently. Despite the prevalence of MV disease (MVD), the cellular and molecular pathways that initiate and perpetuate it are not well understood.
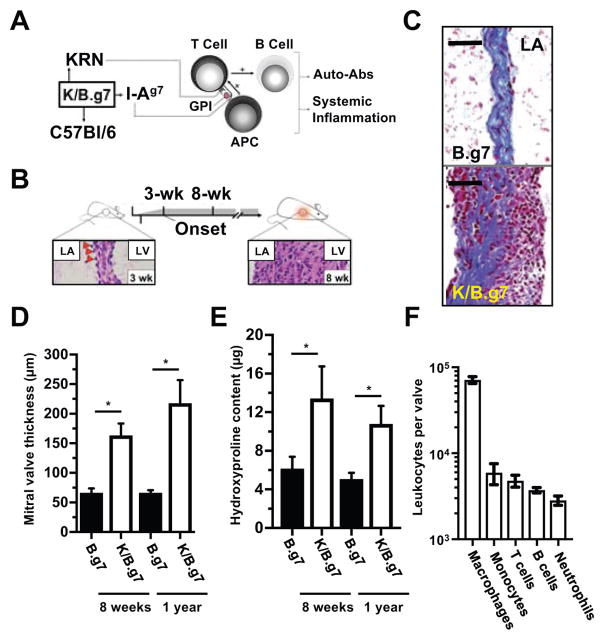
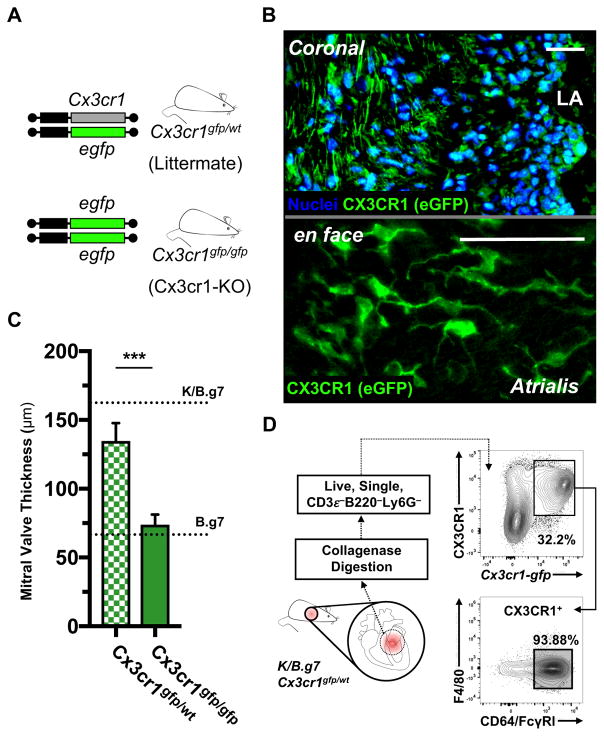
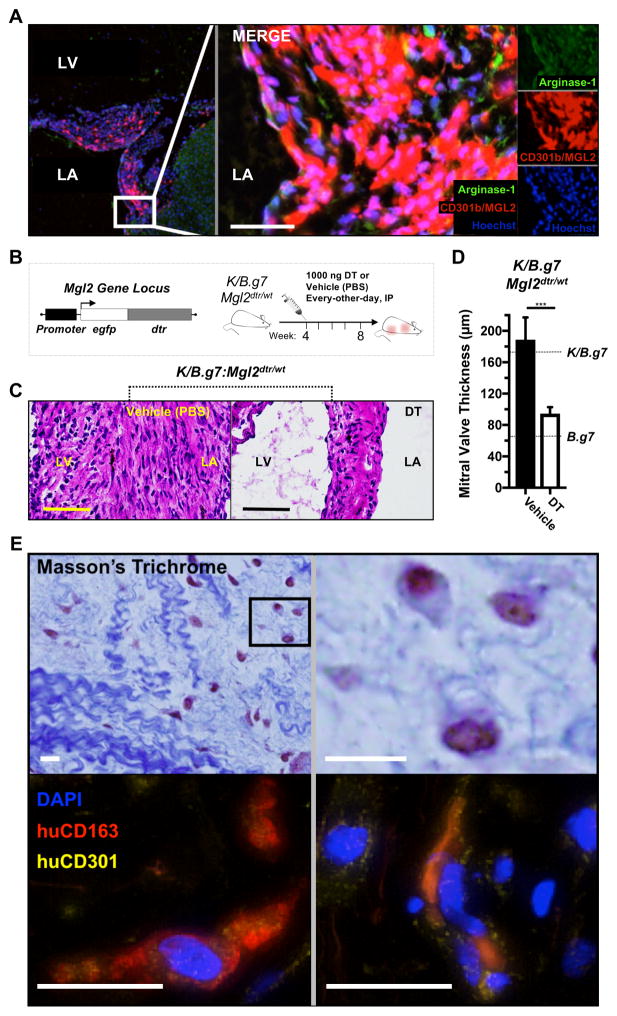
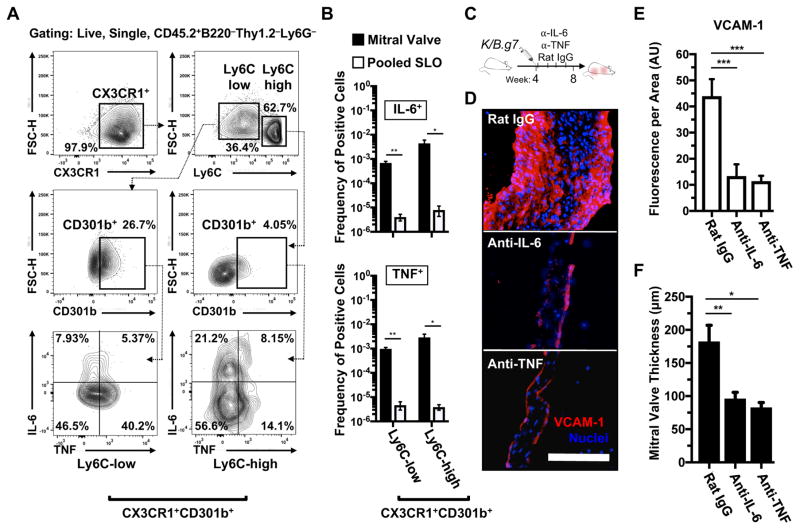
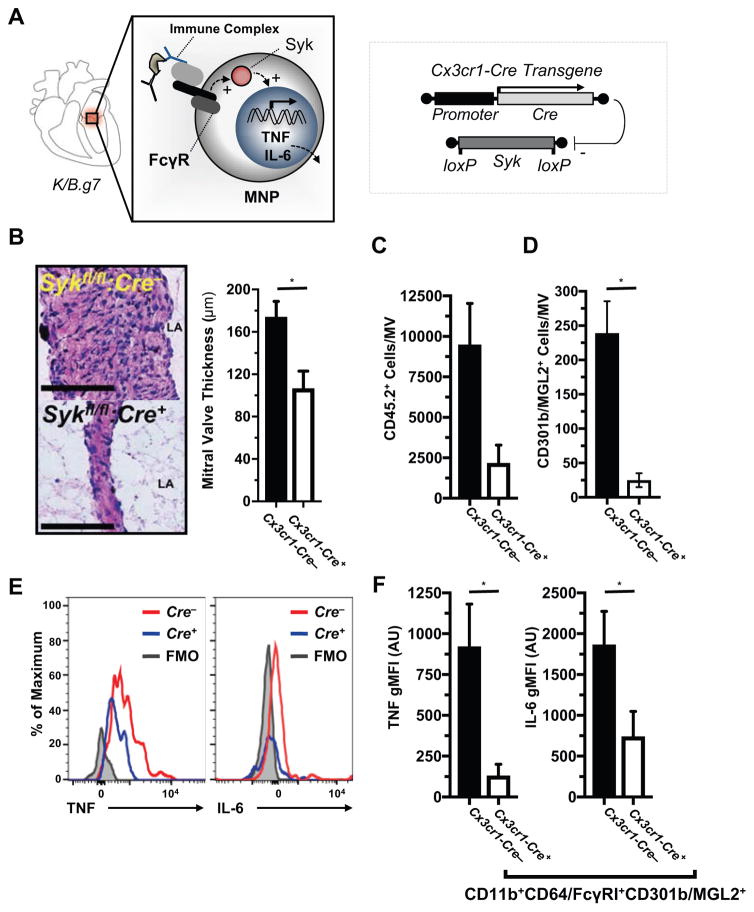
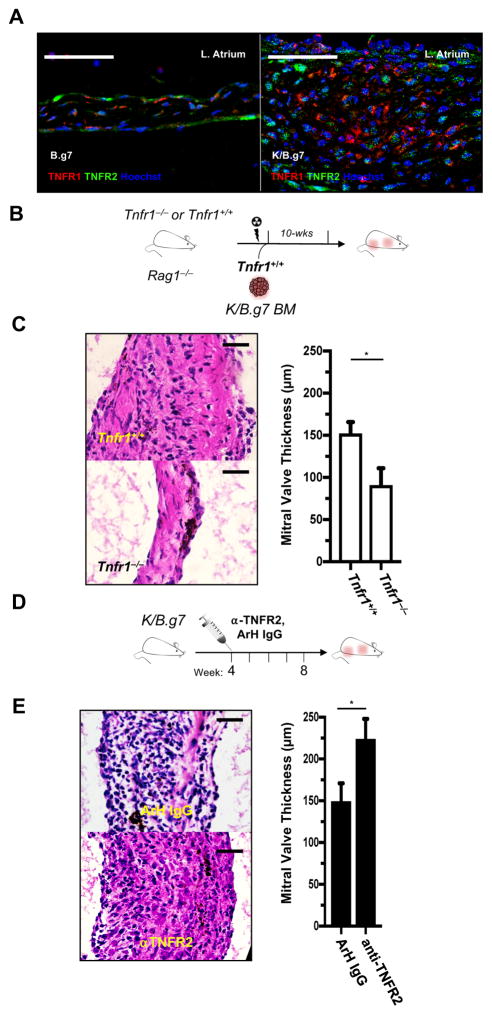
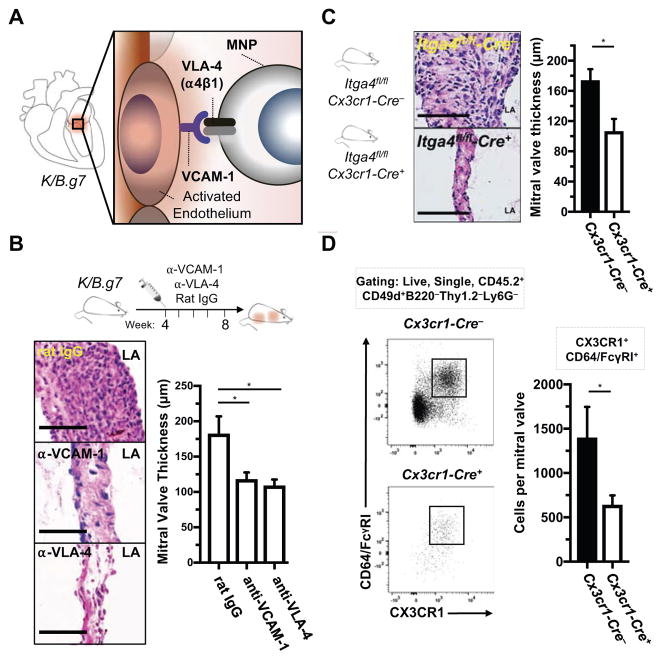
Methods: K/B.g7 T-cell receptor transgenic mice spontaneously develop systemic autoantibody-associated autoimmunity, leading to fully penetrant fibroinflammatory MVD and arthritis. We used multiparameter flow cytometry, intracellular cytokine staining, and immunofluorescent staining to characterize the cells in inflamed K/B.g7 MVs. We used genetic approaches to study the contribution of mononuclear phagocytes (MNPs) to MVD in this model. Specifically, we generated K/B.g7 mice in which either CX3CR1 or CD301b/macrophage galactose N-acetylgalactosamine-specific lectin 2 (MGL2)-expressing MNPs were ablated. Using K/B.g7 mice expressing Cx3Cr1-Cre, we conditionally deleted critical inflammatory molecules from MNPs, including the Fc-receptor signal-transducing tyrosine kinase Syk and the cell adhesion molecule very late antigen-4. We performed complementary studies using monoclonal antibodies to block key inflammatory molecules. We generated bone marrow chimeric mice to define the origin of the inflammatory cells present in the MV and to determine which valve cells respond to the proinflammatory cytokine tumor necrosis factor (TNF). Finally, we examined specimens from patients with rheumatic heart disease to correlate our findings to human pathology.
Results: MNPs comprised the vast majority of MV-infiltrating cells; these MNPs expressed CX3CR1 and CD301b/MGL2. Analogous cells were present in human rheumatic heart disease valves. K/B.g7 mice lacking CX3CR1 or in which CD301b/MGL2-expressing MNPs were ablated were protected from MVD. The valve-infiltrating CD301b/MGL2+ MNPs expressed tissue-reparative molecules including arginase-1 and resistin-like molecule α. These MNPs also expressed the proinflammatory cytokines TNF and interleukin-6, and antibody blockade of these cytokines prevented MVD. Deleting Syk from CX3CR1-expressing MNPs reduced their TNF and interleukin-6 production and also prevented MVD. TNF acted through TNF receptor-1 expressed on valve-resident cells to increase the expression of vascular cell adhesion molecule-1. Conditionally deleting the vascular cell adhesion molecule-1 ligand very late antigen-4 from CX3CR1-expressing MNPs prevented MVD.
Conclusions: CD301b/MGL2+ MNPs are key drivers of autoimmune MVD in K/B.g7 mice and are also present in human rheumatic heart disease. We define key inflammatory molecules that drive MVD in this model, including Syk, TNF, interleukin-6, very late antigen-4, and vascular cell adhesion molecule-1.
Keywords: autoimmune diseases; fibrosis; heart valves; inflammation; macrophages; rheumatic heart disease.
© 2018 American Heart Association, Inc.
Figures

Comment in
-
Myeloid Cells Remodel the Mitral Valve.Circulation. 2018 Jun 5;137(23):2494-2496. doi: 10.1161/CIRCULATIONAHA.118.033622. Circulation. 2018. PMID: 29866774 Free PMC article. No abstract available.
References
-
- Levine RA, Hagege AA, Judge DP, Padala M, Dal-Bianco JP, Aikawa E, Beaudoin J, Bischoff J, Bouatia-Naji N, Bruneval P, Butcher JT, Carpentier A, Chaput M, Chester AH, Clusel C, Delling FN, Dietz HC, Dina C, Durst R, Fernandez-Friera L, Handschumacher MD, Jensen MO, Jeunemaitre XP, Le Marec H, Le Tourneau T, Markwald RR, Merot J, Messas E, Milan DP, Neri T, Norris RA, Peal D, Perrocheau M, Probst V, Puceat M, Rosenthal N, Solis J, Schott JJ, Schwammenthal E, Slaugenhaupt SA, Song JK, Yacoub MH. Mitral valve disease--morphology and mechanisms. Nat Rev Cardiol. 2015;12:689–710. doi: 10.1038/nrcardio.2015.161. - DOI - PMC - PubMed
-
- Nakashima Y, Plump AS, Raines EW, Breslow JL, Ross R. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb. 1994;14:133–140. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
