

Tumour risks and genotype-phenotype correlations associated with germline variants in succinate dehydrogenase subunit genes SDHB, SDHC and SDHD
- PMID: 29386252
- PMCID: PMC5992372
- DOI: 10.1136/jmedgenet-2017-105127
Tumour risks and genotype-phenotype correlations associated with germline variants in succinate dehydrogenase subunit genes SDHB, SDHC and SDHD
Erratum in
-
Correction: Tumour risks and genotype-phenotype correlations associated with germline variants in the succinate dehydrogenase subunit genes SDHB, SDHC, and SDHD.J Med Genet. 2019 Jan;56(1):50-52. doi: 10.1136/jmedgenet-2017-105127corr1. Epub 2018 Nov 22. J Med Genet. 2019. PMID: 30467181 Free PMC article. No abstract available.
Abstract
Background: Germline pathogenic variants in SDHB/SDHC/SDHD are the most frequent causes of inherited phaeochromocytomas/paragangliomas. Insufficient information regarding penetrance and phenotypic variability hinders optimum management of mutation carriers. We estimate penetrance for symptomatic tumours and elucidate genotype-phenotype correlations in a large cohort of SDHB/SDHC/SDHD mutation carriers.
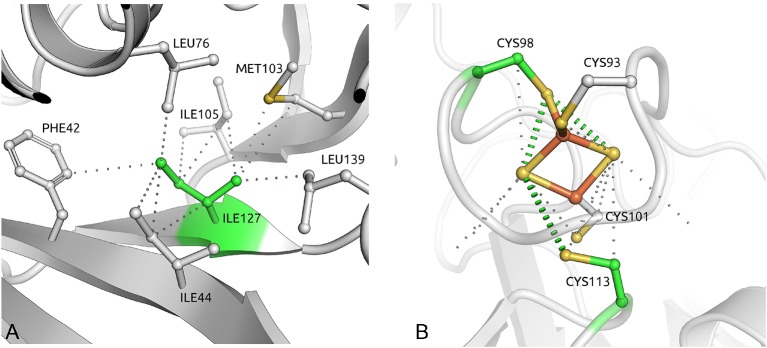
Methods: A retrospective survey of 1832 individuals referred for genetic testing due to a personal or family history of phaeochromocytoma/paraganglioma. 876 patients (401 previously reported) had a germline mutation in SDHB/SDHC/SDHD (n=673/43/160). Tumour risks were correlated with in silico structural prediction analyses.
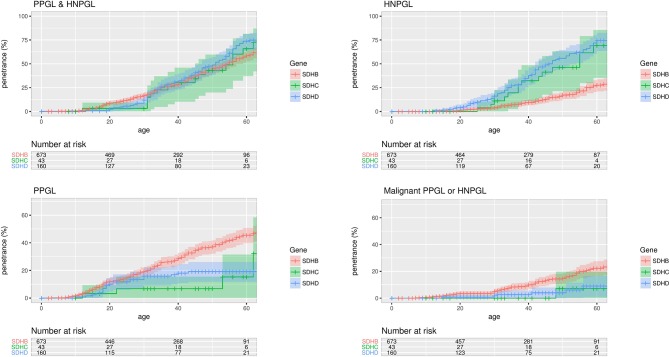
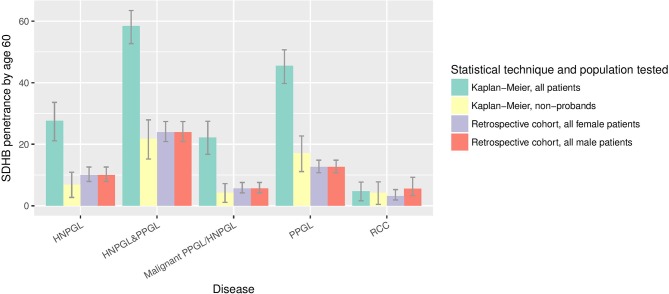
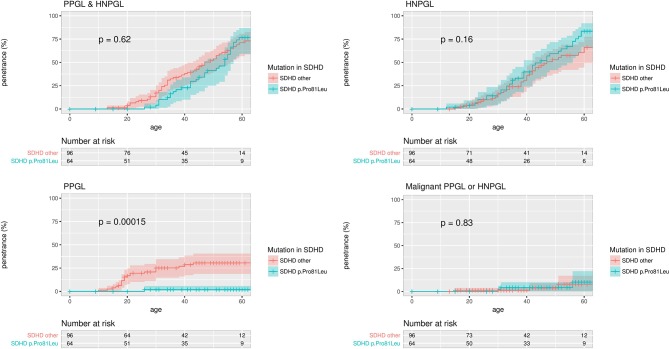
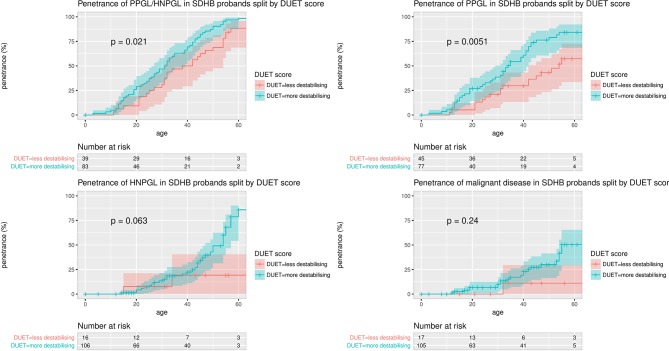
Results: Tumour risks analysis provided novel penetrance estimates and genotype-phenotype correlations. In addition to tumour type susceptibility differences for individual genes, we confirmed that the SDHD:p.Pro81Leu mutation has a distinct phenotype and identified increased age-related tumour risks with highly destabilising SDHB missense mutations. By Kaplan-Meier analysis, the penetrance (cumulative risk of clinically apparent tumours) in SDHB and (paternally inherited) SDHD mutation-positive non-probands (n=371/67 with detailed clinical information) by age 60 years was 21.8% (95% CI 15.2% to 27.9%) and 43.2% (95% CI 25.4% to 56.7%), respectively. Risk of malignant disease at age 60 years in non-proband SDHB mutation carriers was 4.2%(95% CI 1.1% to 7.2%). With retrospective cohort analysis to adjust for ascertainment, cumulative tumour risks for SDHB mutation carriers at ages 60 years and 80 years were 23.9% (95% CI 20.9% to 27.4%) and 30.6% (95% CI 26.8% to 34.7%).
Conclusions: Overall risks of clinically apparent tumours for SDHB mutation carriers are substantially lower than initially estimated and will improve counselling of affected families. Specific genotype-tumour risk associations provides a basis for novel investigative strategies into succinate dehydrogenase-related mechanisms of tumourigenesis and the development of personalised management for SDHB/SDHC/SDHD mutation carriers.
Keywords: cancer: endocrine; genetic epidemiology; genetics; molecular genetics; oncology.
© Article author(s) (or their employer(s) unless otherwise stated in the text of the article) 2018. All rights reserved. No commercial use is permitted unless otherwise expressly granted.
Conflict of interest statement
Competing interests: None declared.
Figures
References
-
- Toledo RA, Burnichon N, Cascon A, Benn DE, Bayley JP, Welander J, Tops CM, Firth H, Dwight T, Ercolino T, Mannelli M, Opocher G, Clifton-Bligh R, Gimm O, Maher ER, Robledo M, Gimenez-Roqueplo AP, Dahia PL; NGS in PPGL (NGSnPPGL) Study Group. Consensus Statement on next-generation-sequencing-based diagnostic testing of hereditary phaeochromocytomas and paragangliomas. Nat Rev Endocrinol 2017;13:233–47. 10.1038/nrendo.2016.185 - DOI - PubMed
-
- Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, van der Mey A, Taschner PE, Rubinstein WS, Myers EN, Richard CW, Cornelisse CJ, Devilee P, Devlin B. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 2000;287:848–51. 10.1126/science.287.5454.848 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical