

Hexokinase-2 depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin
- PMID: 29386513
- PMCID: PMC5792493
- DOI: 10.1038/s41467-017-02733-4
Hexokinase-2 depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin
Erratum in
-
Author Correction: Hexokinase-2 depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin.Nat Commun. 2018 Jun 26;9(1):2539. doi: 10.1038/s41467-018-04182-z. Nat Commun. 2018. PMID: 29946147 Free PMC article.
Abstract
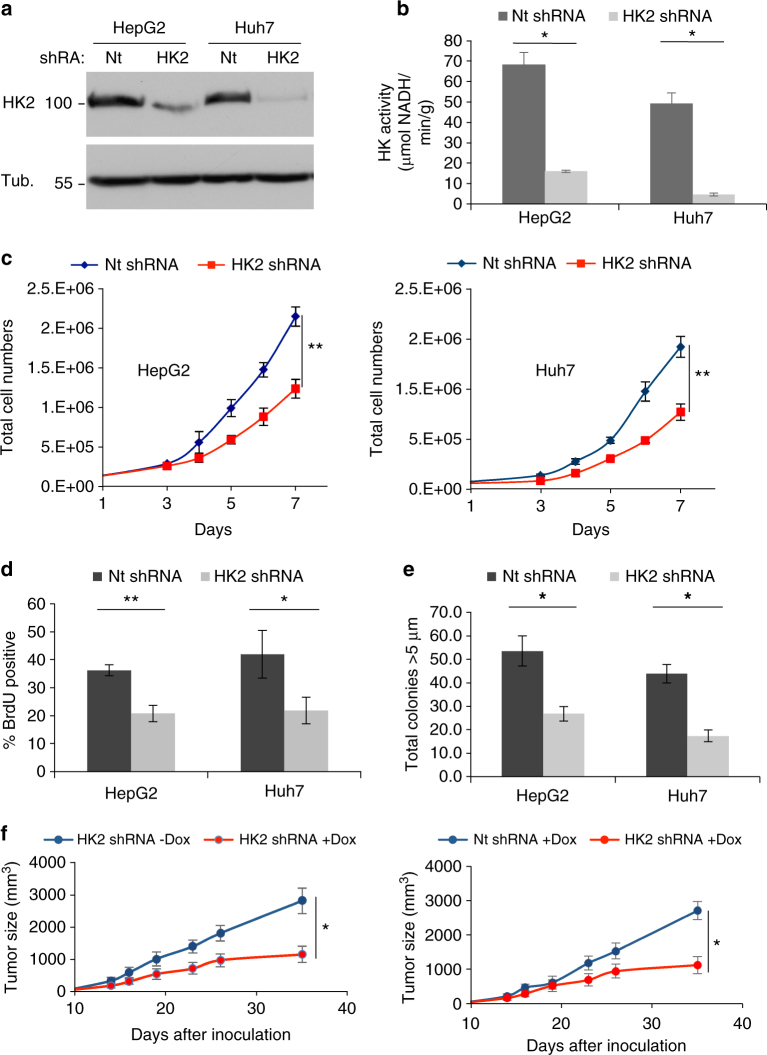
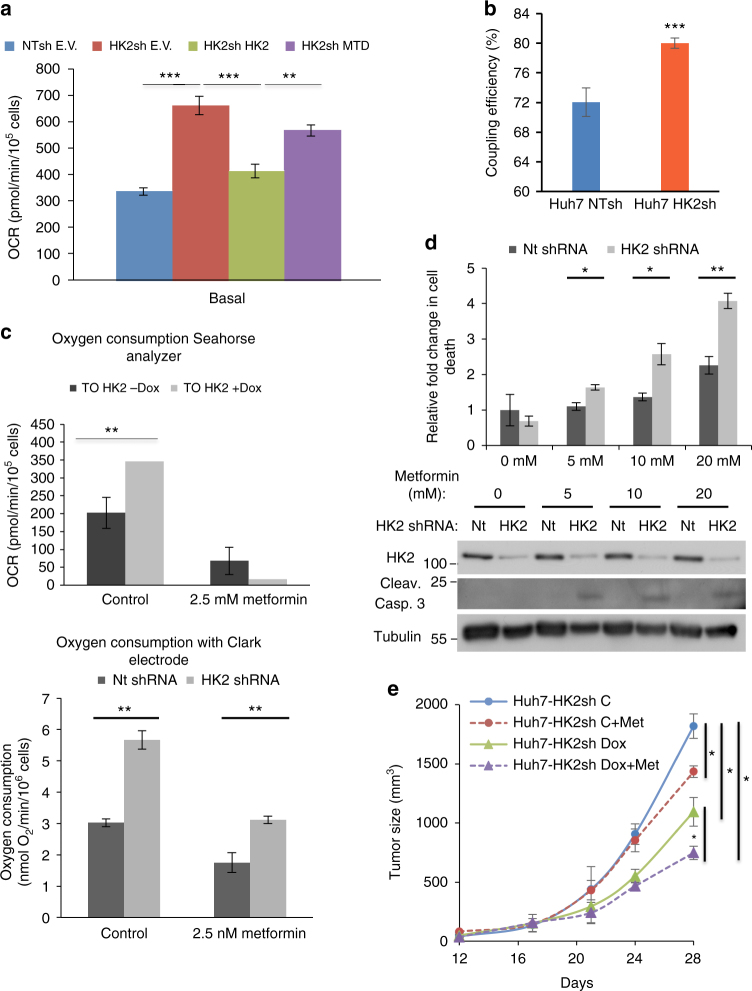
Hepatocellular carcinoma (HCC) cells are metabolically distinct from normal hepatocytes by expressing the high-affinity hexokinase (HK2) and suppressing glucokinase (GCK). This is exploited to selectively target HCC. Hepatic HK2 deletion inhibits tumor incidence in a mouse model of hepatocarcinogenesis. Silencing HK2 in human HCC cells inhibits tumorigenesis and increases cell death, which cannot be restored by GCK or mitochondrial binding deficient HK2. Upon HK2 silencing, glucose flux to pyruvate and lactate is inhibited, but TCA fluxes are maintained. Serine uptake and glycine secretion are elevated suggesting increased requirement for one-carbon contribution. Consistently, vulnerability to serine depletion increases. The decrease in glycolysis is coupled to elevated oxidative phosphorylation, which is diminished by metformin, further increasing cell death and inhibiting tumor growth. Neither HK2 silencing nor metformin alone inhibits mTORC1, but their combination inhibits mTORC1 in an AMPK-independent and REDD1-dependent mechanism. Finally, HK2 silencing synergizes with sorafenib to inhibit tumor growth.
Conflict of interest statement
The authors declare no competing financial interests.
Figures






References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
