

Clinical heterogeneity of mitochondrial NAD kinase deficiency caused by a NADK2 start loss variant
- PMID: 29388319
- PMCID: PMC6185736
- DOI: 10.1002/ajmg.a.38602
Clinical heterogeneity of mitochondrial NAD kinase deficiency caused by a NADK2 start loss variant
Abstract
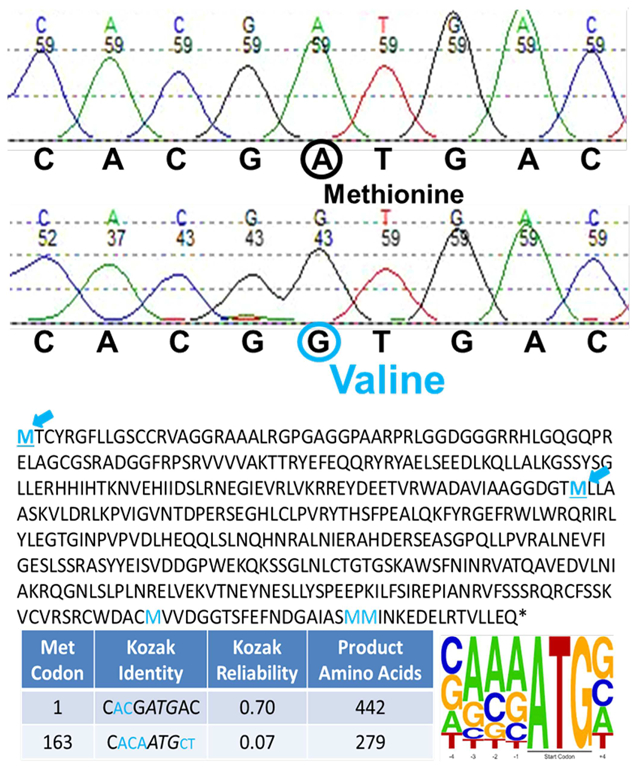
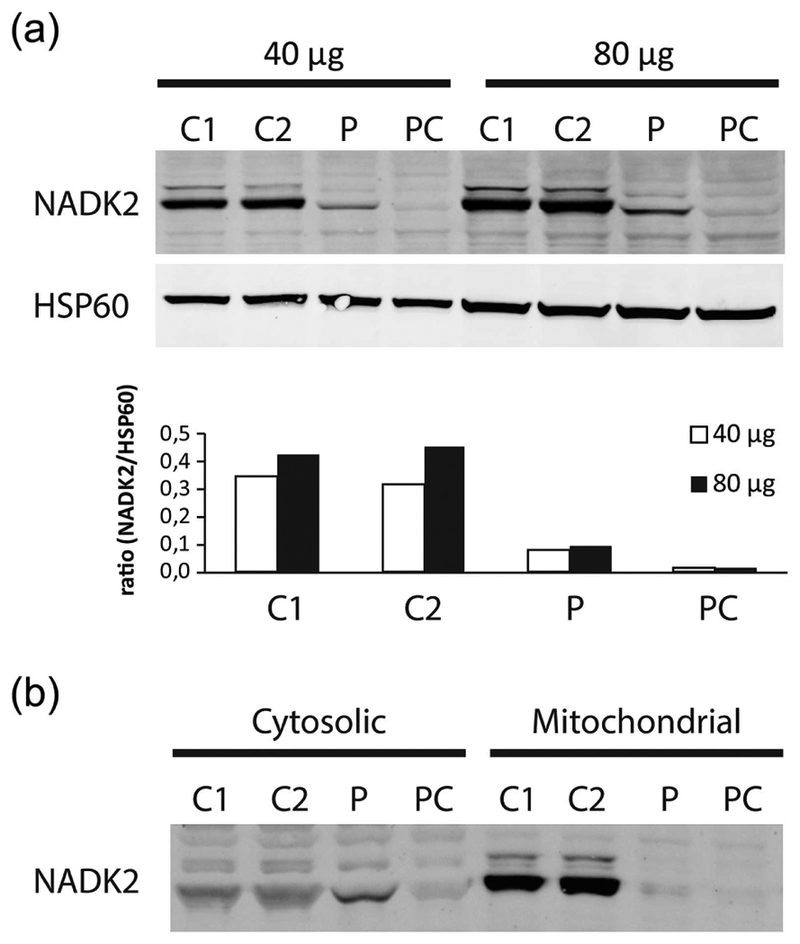
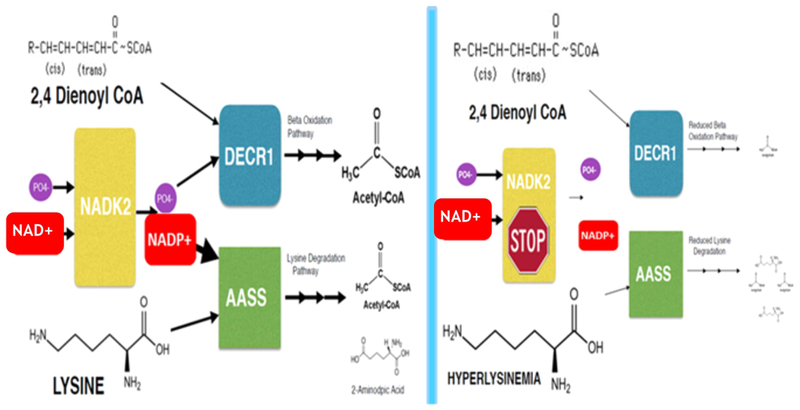
Mitochondrial NAD kinase deficiency (NADK2D, OMIM #615787) is a rare autosomal recessive disorder of NADPH biosynthesis that can cause hyperlysinemia and dienoyl-CoA reductase deficiency (DECRD, OMIM #616034). NADK2 deficiency has been reported in only three unrelated patients. Two had severe, unremitting disease; one died at 4 months and the other at 5 years of age. The third was a 10 year old female with CNS anomalies, ataxia, and incoordination. In two cases mutations in NADK2 have been demonstrated. Here, we report the fourth known case, a 15 year old female with normal intelligence and a mild clinical and biochemical phenotype presumably without DECRD. Her clinical symptoms, which are now stable, became evident at the age of 9 with the onset of decreased visual acuity, bilateral optic atrophy, nystagmus, episodic lower extremity weakness, peripheral neuropathy, and gait abnormalities. Plasma amino acid levels were within normal limits except for mean lysine and proline levels that were 3.7 and 2.5 times the upper limits of normal. Whole exome sequencing (WES) revealed homozygosity for a g.36241900 A>G p. Met1Val start loss mutation in the primary NADK2 transcript (NM_001085411.1) encoding the 442 amino acid isoform. This presumed hypomorphic mutation has not been previously reported and is absent from the v1000GP, EVS, and ExAC databases. Our patient's normal intelligence and stable disease expands the clinical heterogeneity and the prognosis associated with NADK2 deficiency. Our findings also clarify the mechanism underlying NADK2 deficiency and suggest that this disease should be ruled out in cases of hyperlysinemia, especially those with visual loss, and neurological phenotypes.
Keywords: 2,4 dienoyl-CoA reductase deficiency; NADK2; NADPH; hyperlysinemia; optic atrophy; start-loss codon.
© 2018 Wiley Periodicals, Inc.
Figures
References
-
- Calva-Cerqueira D, Chinnathambi S, Pechman B, Bair J, Larsen-Haidle J, & Howe JR (2009). The Rate of germline mutations and large deletions of SMAD4 and BMPR1A in juvenile polyposis. Clinical Genetics, 75(1), 79–85. - PubMed
-
- Felgentreff K, Perez-Becker R, Speckmann C, Schwarz K, Kalwak K, Markelj G, … Ehl S (2011). Clinical and immunological manifestations of patients with atypical severe combined immunodeficiency. Clinical Immunology, 141(1), 73–82. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
