

Infantile-onset Pompe disease with neonatal debut: A case report and literature review
- PMID: 29390460
- PMCID: PMC5758162
- DOI: 10.1097/MD.0000000000009186
Infantile-onset Pompe disease with neonatal debut: A case report and literature review
Abstract
Rationale: Infantile-onset Pompe disease, also known as glycogen storage disease type II, is a progressive and fatal disorder without treatment. Enzyme replacement therapy with recombinant human acid alpha-glucosidase (GAA) enhances survival; however, the best outcomes have been achieved with early treatment.
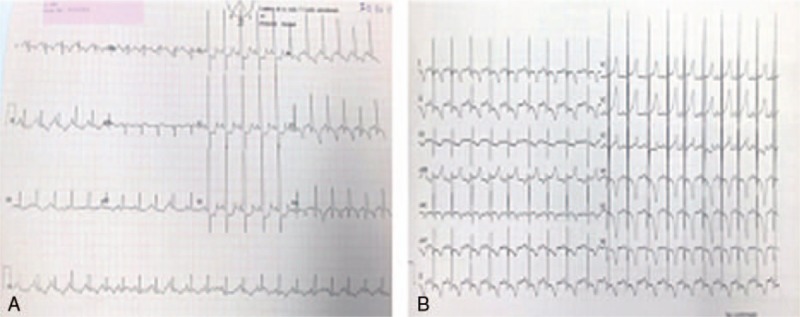
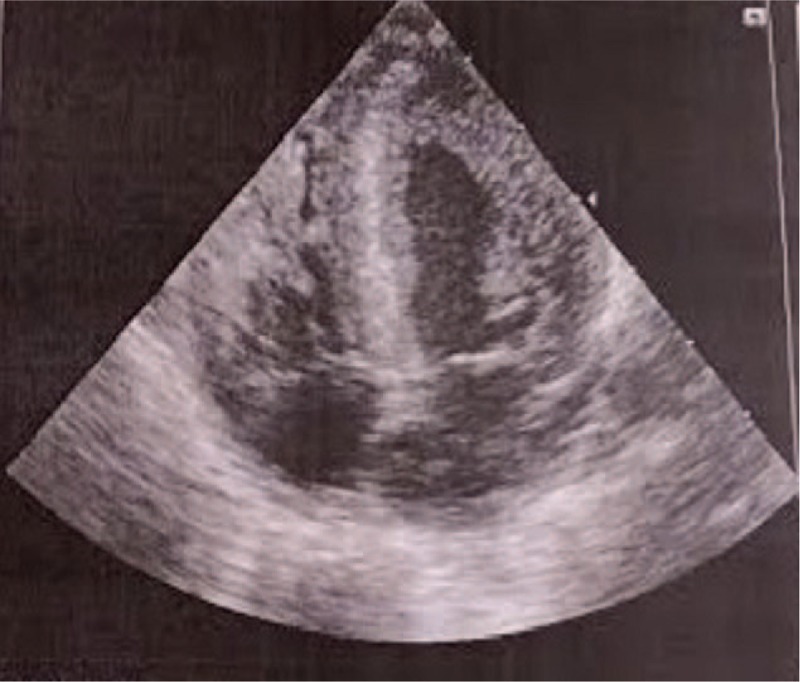
Patient concerns: We report a case of a newborn with infantile-onset Pompe disease diagnosed in the first days of life who did not undergo universal neonatal screening. The patient was asymptomatic, with a general physical examination revealing only a murmur. The clinical presentation was dominated by the neonatal detection of hypertrophic cardiomyopathy, without hypotonia or macroglossia.
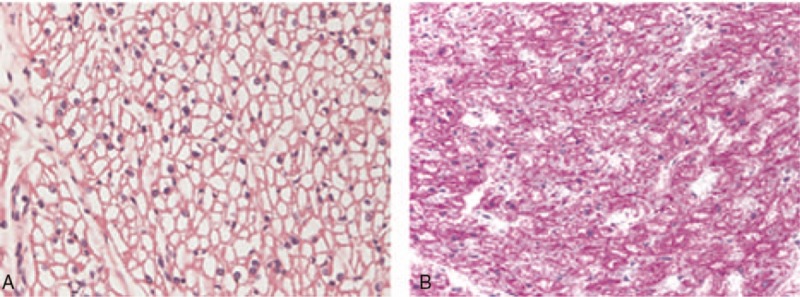
Diagnoses: Pompe disease was confirmed in the first week of life by GAA activity in dried blood spots, and a GAA genetic study showed the homozygous mutation p.Arg854X.
Interventions: Parents initially refused replacement therapy.
Outcomes: The patient experienced recurrent episodes of ventricular fibrillation during central line placement and could not be resuscitated.
Lessons: Although Pompe disease is rare, and universal screening has not been established, neonatologists should be alerted to the diagnosis of Pompe in the presence of hypertrophic cardiomyopathy. Diagnosis is achieved in a few days with the aid of dried blood spots.
Copyright © 2017 The Authors. Published by Wolters Kluwer Health, Inc. All rights reserved.
Conflict of interest statement
The authors have no conflicts of interest to disclose.
Figures
References
-
- Lawrence Merrit J. Lysosomal acid alpha-glucosidase deficiency (Pompe disease, glycogen storage disease II, acid maltase deficiency). Rose BD ed. Waltham, MA, UpToDate, 2005. Available at: http://www.uptodate.com/. Accessed January 30, 2016.
-
- Van der Ploeg AT, Reuser AJJ. Pompe's disease. Lancet 2008;372:1342–53. - PubMed
-
- Pascual-Pascual SI, Nascimento A, Fernandez-Llamazares CM, et al. Clinical guidelines for infantile-onset Pompe disease. Rev Neurol 2016;63:269–79. - PubMed
-
- Elliott PM, Anastasakis A, Borger MA, et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy. The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 2014;35:2733–79. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
