

Human ADAR1 Prevents Endogenous RNA from Triggering Translational Shutdown
- PMID: 29395325
- PMCID: PMC5831367
- DOI: 10.1016/j.cell.2017.12.038
Human ADAR1 Prevents Endogenous RNA from Triggering Translational Shutdown
Abstract
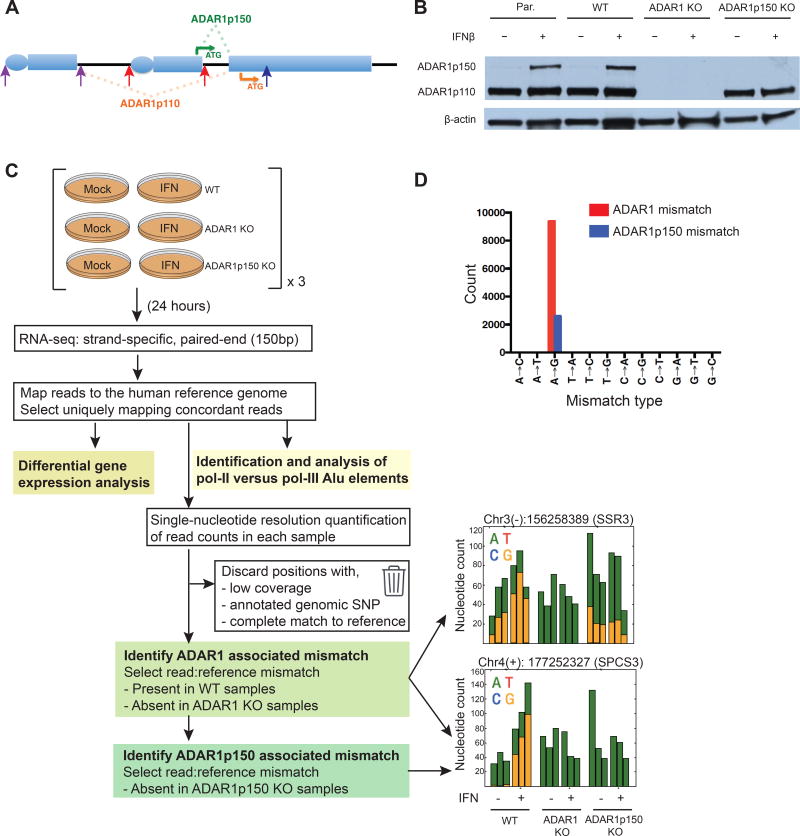
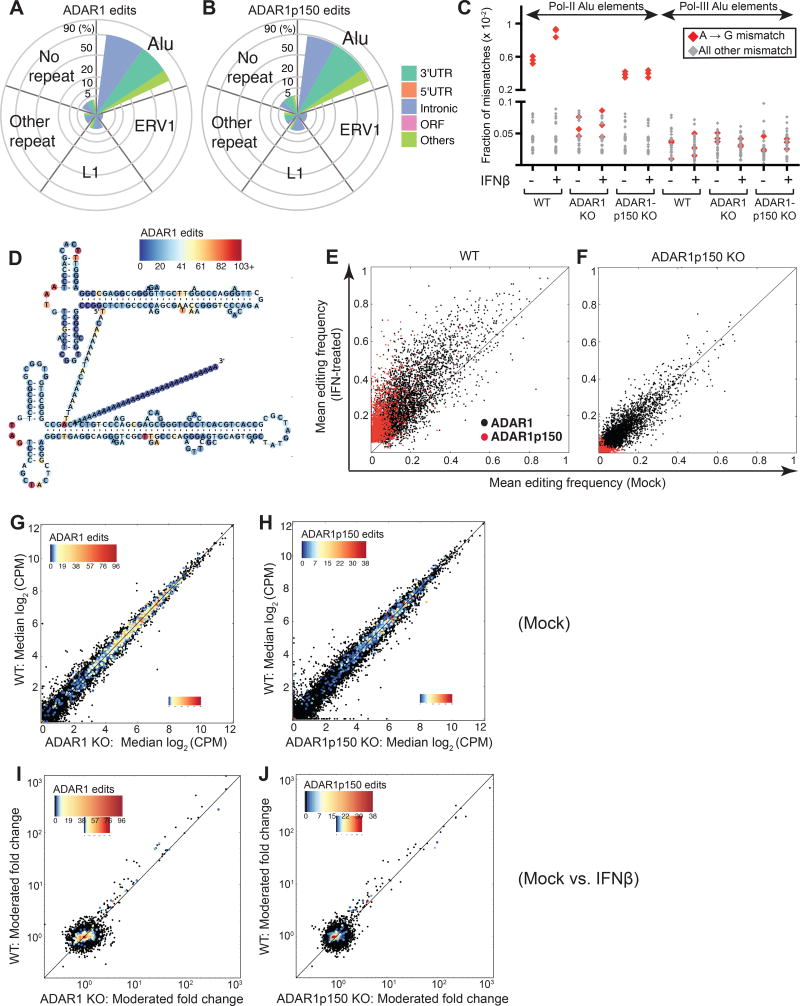
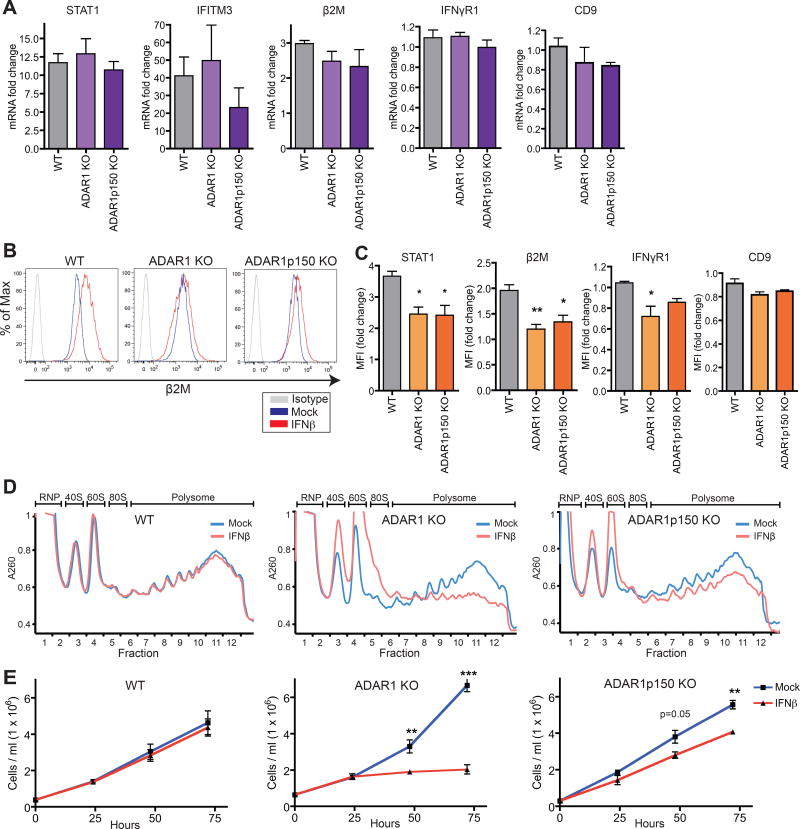
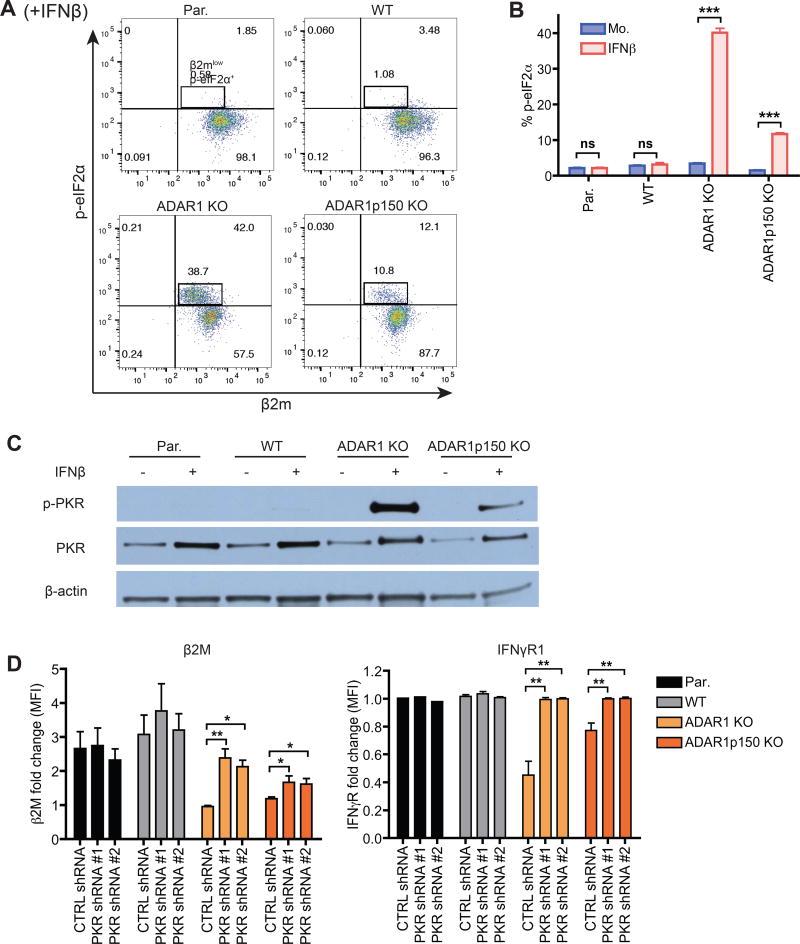
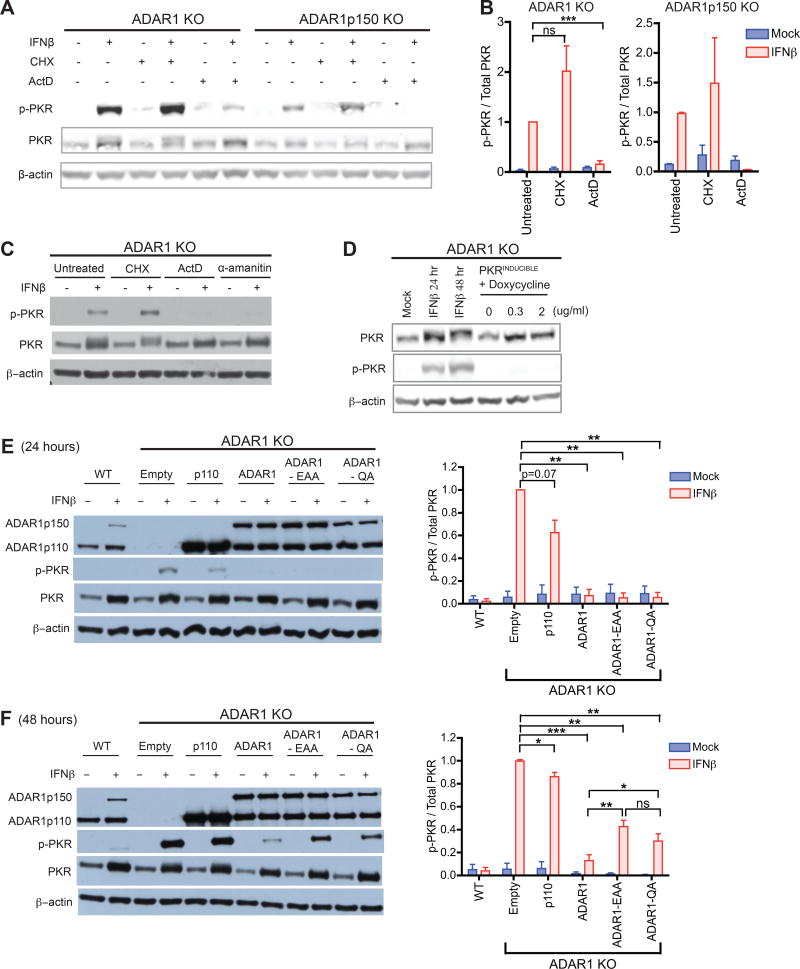
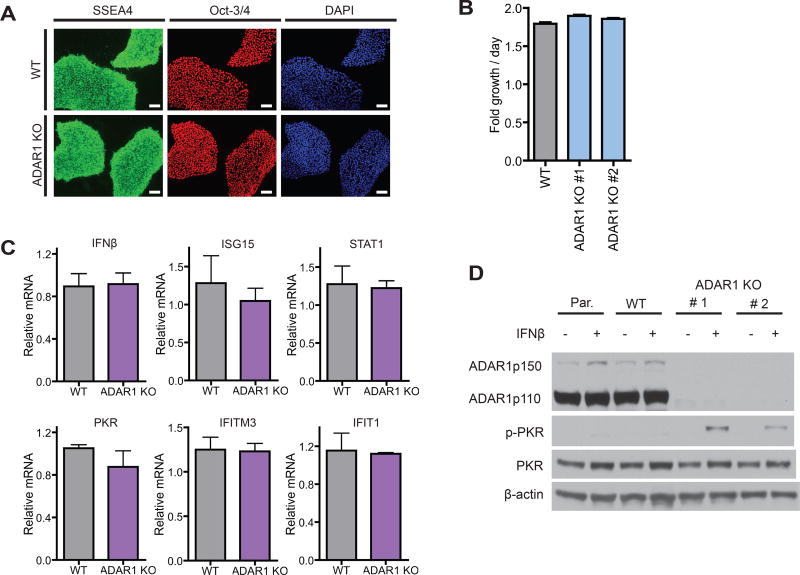
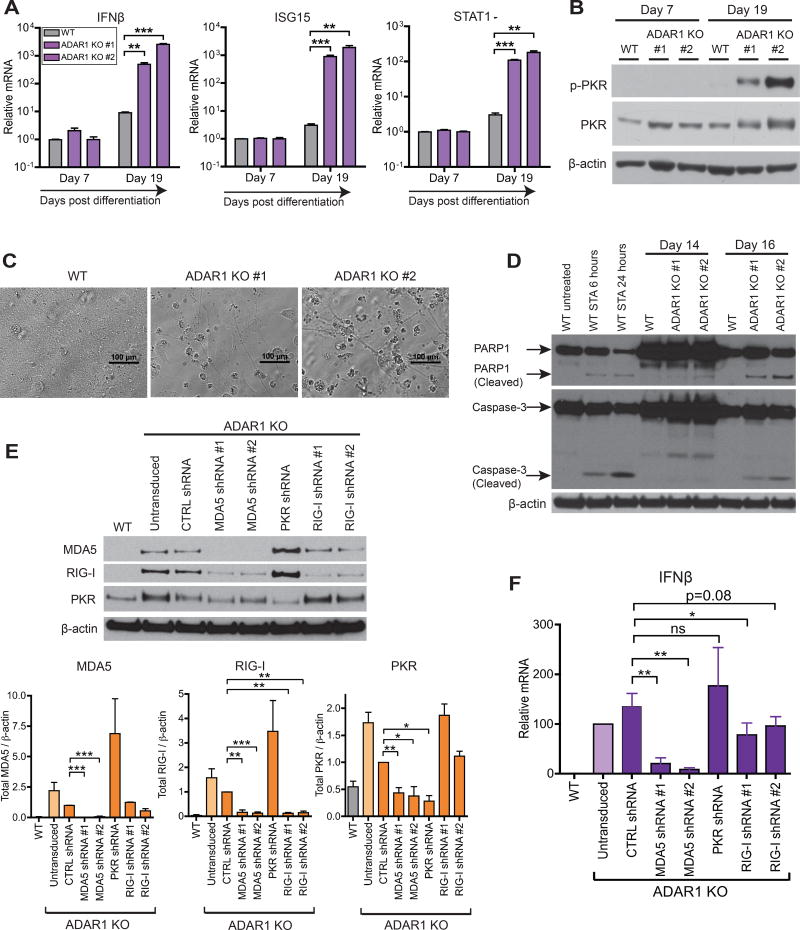
Type I interferon (IFN) is produced when host sensors detect foreign nucleic acids, but how sensors differentiate self from nonself nucleic acids, such as double-stranded RNA (dsRNA), is incompletely understood. Mutations in ADAR1, an adenosine-to-inosine editing enzyme of dsRNA, cause Aicardi-Goutières syndrome, an autoinflammatory disorder associated with spontaneous interferon production and neurologic sequelae. We generated ADAR1 knockout human cells to explore ADAR1 substrates and function. ADAR1 primarily edited Alu elements in RNA polymerase II (pol II)-transcribed mRNAs, but not putative pol III-transcribed Alus. During the IFN response, ADAR1 blocked translational shutdown by inhibiting hyperactivation of PKR, a dsRNA sensor. ADAR1 dsRNA binding and catalytic activities were required to fully prevent endogenous RNA from activating PKR. Remarkably, ADAR1 knockout neuronal progenitor cells exhibited MDA5 (dsRNA sensor)-dependent spontaneous interferon production, PKR activation, and cell death. Thus, human ADAR1 regulates sensing of self versus nonself RNA, allowing pathogen detection while avoiding autoinflammation.
Keywords: ADAR1; AGS; Aicardi-Goutieres syndrome; Alu elements; MDA5; PKR; RNA editing; innate immunity; neuronal progenitor cells; translation; type I interferon.
Copyright © 2018 Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare no competing interests.
Figures
Comment in
-
RNA metabolism: Interfering with interferon by RNA editing.Nat Rev Mol Cell Biol. 2018 Mar;19(3):140-141. doi: 10.1038/nrm.2018.12. Epub 2018 Feb 7. Nat Rev Mol Cell Biol. 2018. PMID: 29410527 No abstract available.
-
Sort Your Self Out!Cell. 2018 Feb 8;172(4):640-642. doi: 10.1016/j.cell.2018.01.023. Cell. 2018. PMID: 29425484
-
Who Defends the Stem Cell's Citadel?Cell Stem Cell. 2018 Mar 1;22(3):287-289. doi: 10.1016/j.stem.2018.02.007. Cell Stem Cell. 2018. PMID: 29499145
References
-
- Bass BL, Weintraub H. An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell. 1988;55:1089–1098. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
