

The Value of Genetic Testing in Polycystic Kidney Diseases Illustrated by a Family With PKD2 and COL4A1 Mutations
- PMID: 29395486
- PMCID: PMC6057824
- DOI: 10.1053/j.ajkd.2017.11.015
The Value of Genetic Testing in Polycystic Kidney Diseases Illustrated by a Family With PKD2 and COL4A1 Mutations
Abstract
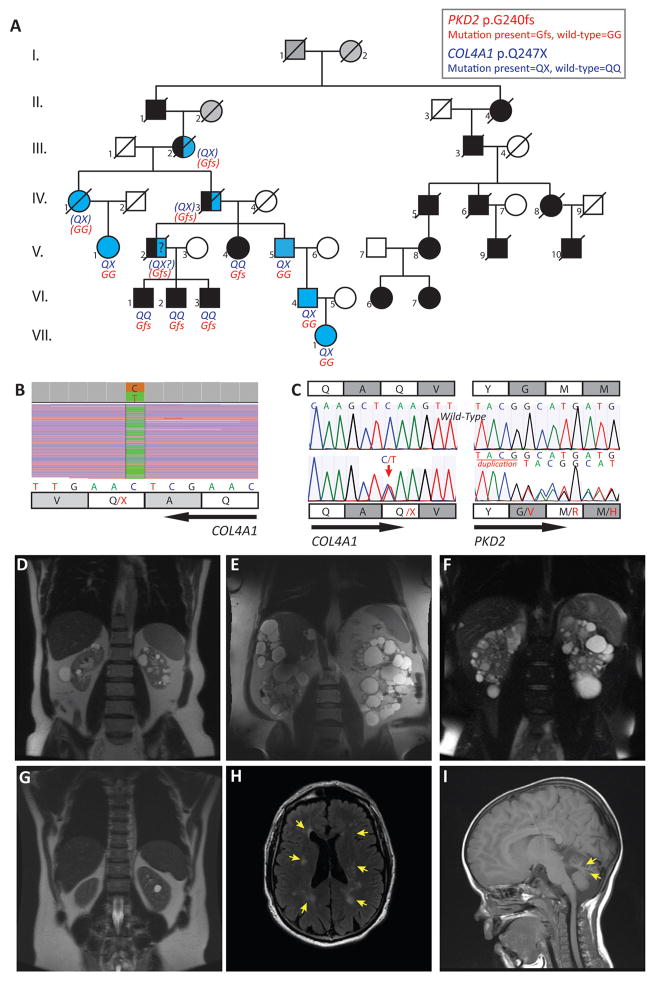
The diagnosis of autosomal dominant polycystic kidney disease (ADPKD) relies on imaging criteria in the setting of a positive familial history. Molecular analysis, seldom used in clinical practice, identifies a causative mutation in >90% of cases in the genes PKD1, PKD2, or rarely GANAB. We report the clinical and genetic dissection of a 7-generation pedigree, resulting in the diagnosis of 2 different cystic disorders. Using targeted next-generation sequencing of 65 candidate genes in a patient with an ADPKD-like phenotype who lacked the familial PKD2 mutation, we identified a COL4A1 mutation (p.Gln247*) and made the diagnosis of HANAC (hereditary angiopathy with nephropathy, aneurysms, and muscle cramps) syndrome. While 4 individuals had ADPKD-PKD2, various COL4A1-related phenotypes were identified in 5 patients, and 3 individuals with likely digenic PKD2/COL4A1 disease reached end-stage renal disease at around 50 years of age, significantly earlier than observed for either monogenic disorder. Thus, using targeted next-generation sequencing as part of the diagnostic approach in patients with cystic diseases provides differential diagnoses and identifies factors underlying disease variability. As specific therapies are rapidly developing for ADPKD, a precise etiologic diagnosis should be paramount for inclusion in therapeutic trials and optimal patient management.
Keywords: Autosomal dominant polycystic kidney disease (ADPKD); COL4A1; HANAC; PKD2; case report; genetic testing; pedigree; phenotypic variability; targeted next-generation sequencing (TNGS).
Copyright © 2017 National Kidney Foundation, Inc. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Ong AC, Devuyst O, Knebelmann B, Walz G Diseases E-EWGfIK. Autosomal dominant polycystic kidney disease: the changing face of clinical management. Lancet. 2015;385:1993–2002. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- U01 DK062401/DK/NIDDK NIH HHS/United States
- R01 DK113111/DK/NIDDK NIH HHS/United States
- P30 DK090728/DK/NIDDK NIH HHS/United States
- U01 DK062402/DK/NIDDK NIH HHS/United States
- P30 DK106912/DK/NIDDK NIH HHS/United States
- U01 DK062408/DK/NIDDK NIH HHS/United States
- U01 DK062411/DK/NIDDK NIH HHS/United States
- U01 DK082230/DK/NIDDK NIH HHS/United States
- U01 DK062410/DK/NIDDK NIH HHS/United States
- R01 DK058816/DK/NIDDK NIH HHS/United States
- R01 DK044863/DK/NIDDK NIH HHS/United States
- UL1 TR000170/TR/NCATS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
