

Stress-Induced Mutagenesis: Implications in Cancer and Drug Resistance
- PMID: 29399660
- PMCID: PMC5794033
- DOI: 10.1146/annurev-cancerbio-050216-121919
Stress-Induced Mutagenesis: Implications in Cancer and Drug Resistance
Abstract
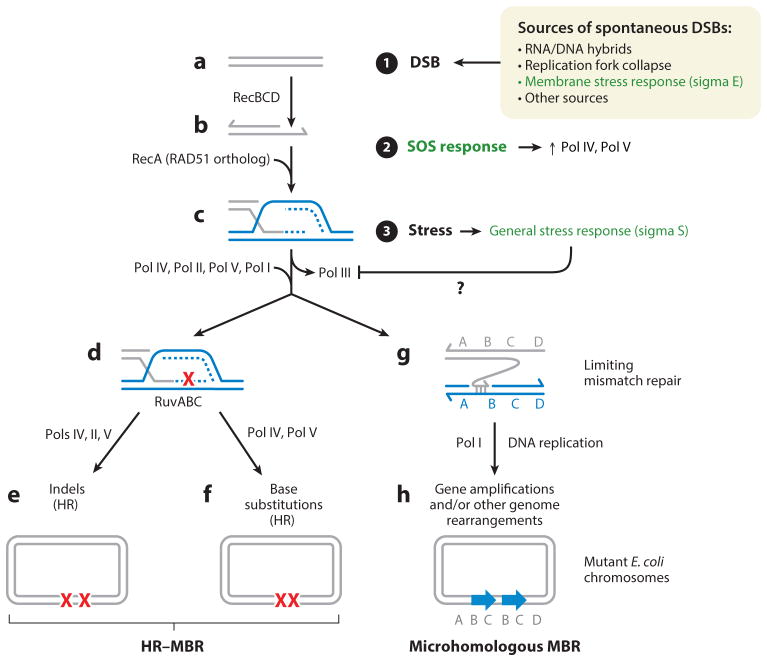
Genomic instability underlies many cancers and generates genetic variation that drives cancer initiation, progression, and therapy resistance. In contrast with classical assumptions that mutations occur purely stochastically at constant, gradual rates, microbes, plants, flies, and human cancer cells possess mechanisms of mutagenesis that are upregulated by stress responses. These generate transient, genetic-diversity bursts that can propel evolution, specifically when cells are poorly adapted to their environments-that is, when stressed. We review molecular mechanisms of stress-response-dependent (stress-induced) mutagenesis that occur from bacteria to cancer, and are activated by starvation, drugs, hypoxia, and other stressors. We discuss mutagenic DNA break repair in Escherichia coli as a model for mechanisms in cancers. The temporal regulation of mutagenesis by stress responses and spatial restriction in genomes are common themes across the tree of life. Both can accelerate evolution, including the evolution of cancers. We discuss possible anti-evolvability drugs, aimed at targeting mutagenesis and other variation generators, that could be used to delay the evolution of cancer progression and therapy resistance.
Keywords: HSP90; MMBIR; chemotherapy; double-strand break repair; evolution; genome instability; hypoxia; kataegis; stress response; trinucleotide repeat instability.
Figures

References
-
- Andersson DI, Hughes D. Microbiological effects of sublethal levels of antibiotics. Nat Rev Microbiol. 2014;12:465–78. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources