

Recent Advances in Detecting Mitochondrial DNA Heteroplasmic Variations
- PMID: 29401641
- PMCID: PMC6017848
- DOI: 10.3390/molecules23020323
Recent Advances in Detecting Mitochondrial DNA Heteroplasmic Variations
Abstract
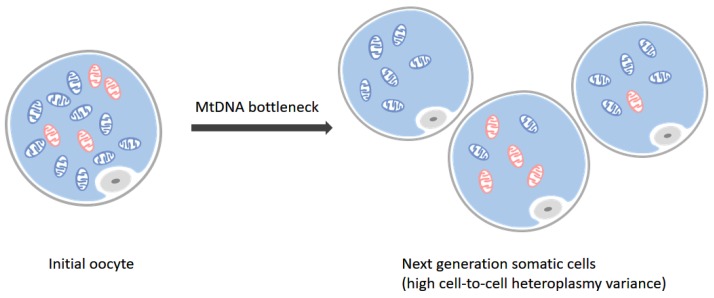
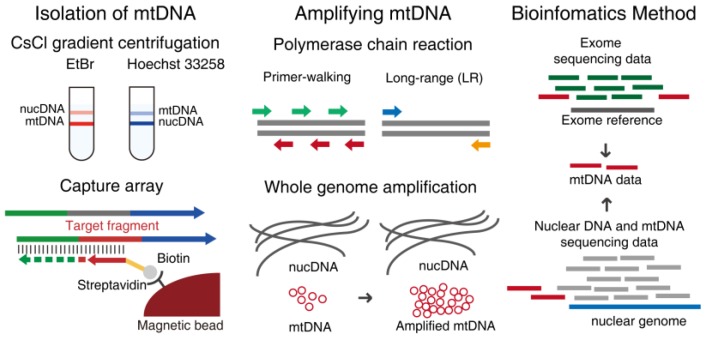
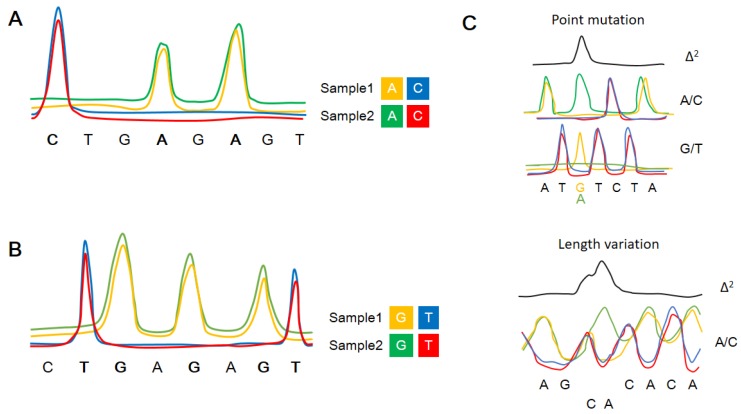
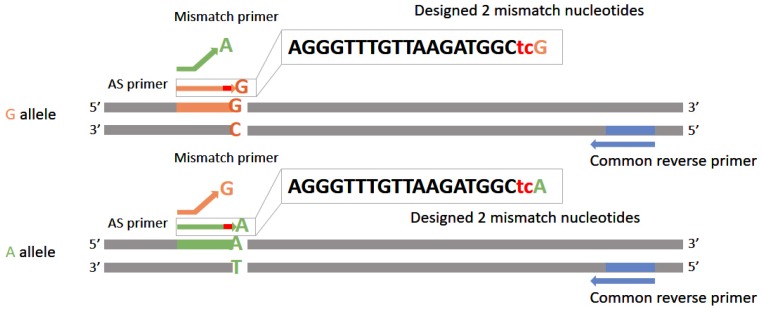
The co-existence of wild-type and mutated mitochondrial DNA (mtDNA) molecules termed heteroplasmy becomes a research hot point of mitochondria. In this review, we listed several methods of mtDNA heteroplasmy research, including the enrichment of mtDNA and the way of calling heteroplasmic variations. At the present, while calling the novel ultra-low level heteroplasmy, high-throughput sequencing method is dominant while the detection limit of recorded mutations is accurate to 0.01% using the other quantitative approaches. In the future, the studies of mtDNA heteroplasmy may pay more attention to the single-cell level and focus on the linkage of mutations.
Keywords: heteroplasmy; mitochondria; mtDNA; next generation sequencing.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
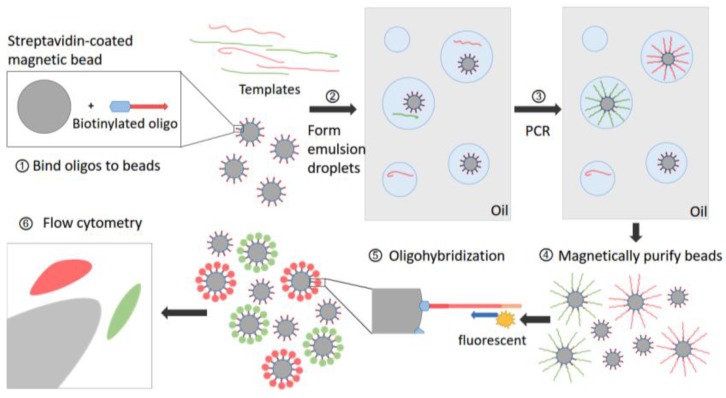
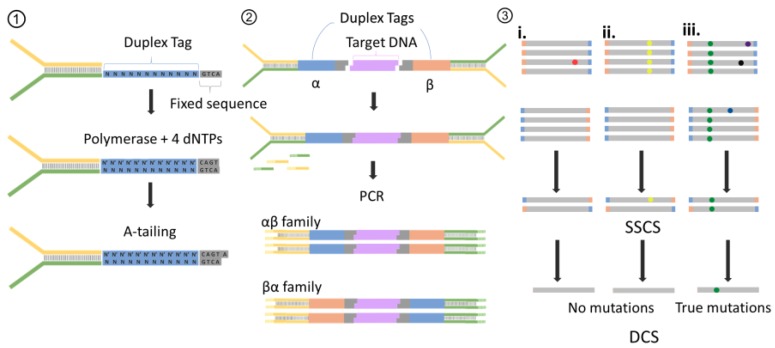
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
