

Defiant: (DMRs: easy, fast, identification and ANnoTation) identifies differentially Methylated regions from iron-deficient rat hippocampus
- PMID: 29402210
- PMCID: PMC5800085
- DOI: 10.1186/s12859-018-2037-1
Defiant: (DMRs: easy, fast, identification and ANnoTation) identifies differentially Methylated regions from iron-deficient rat hippocampus
Abstract
Background: Identification of differentially methylated regions (DMRs) is the initial step towards the study of DNA methylation-mediated gene regulation. Previous approaches to call DMRs suffer from false prediction, use extreme resources, and/or require library installation and input conversion.
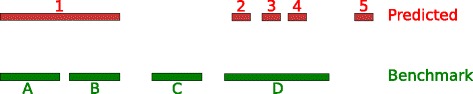
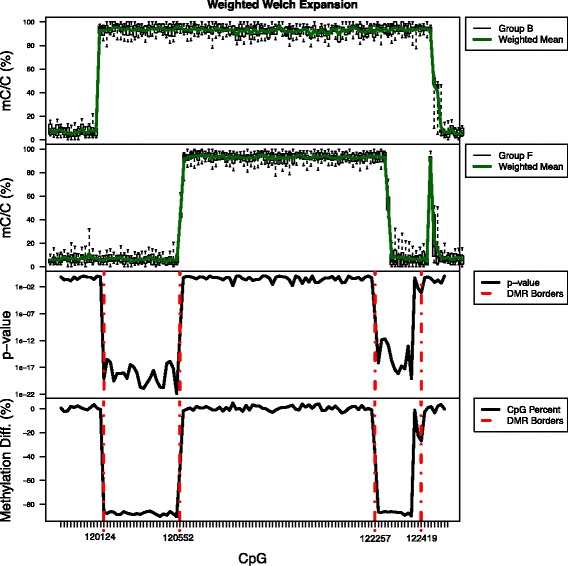
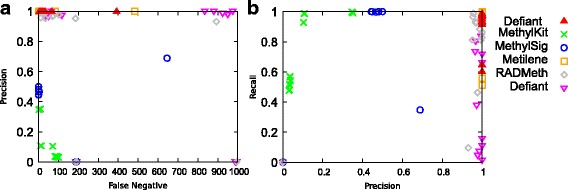
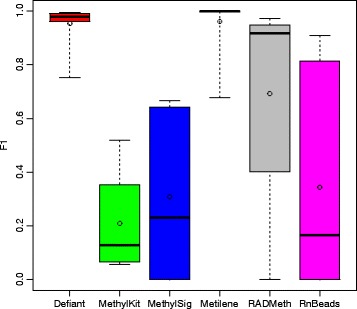
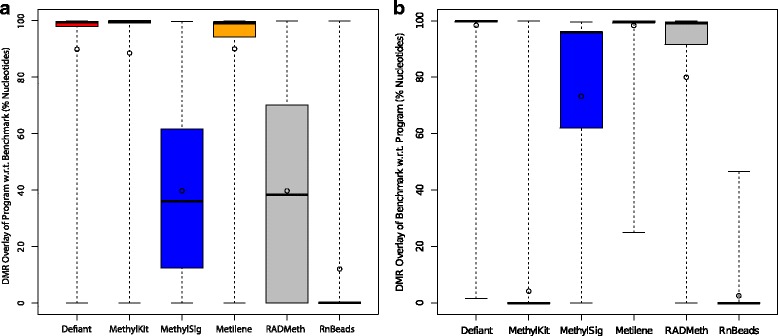
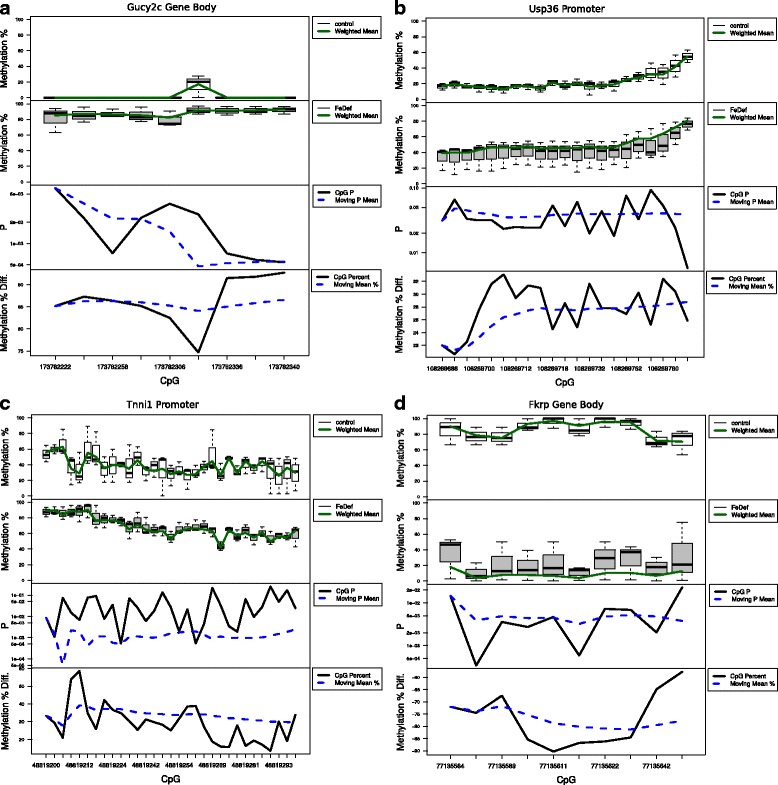
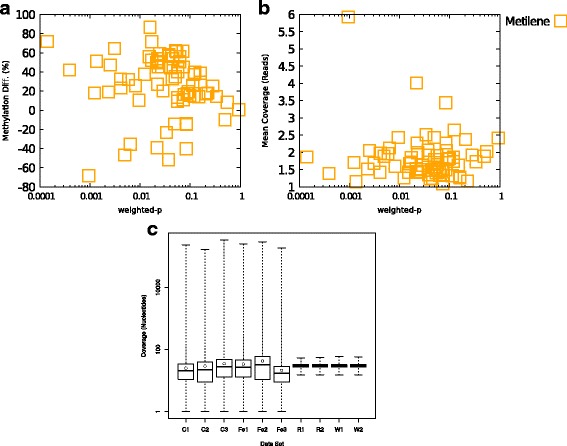
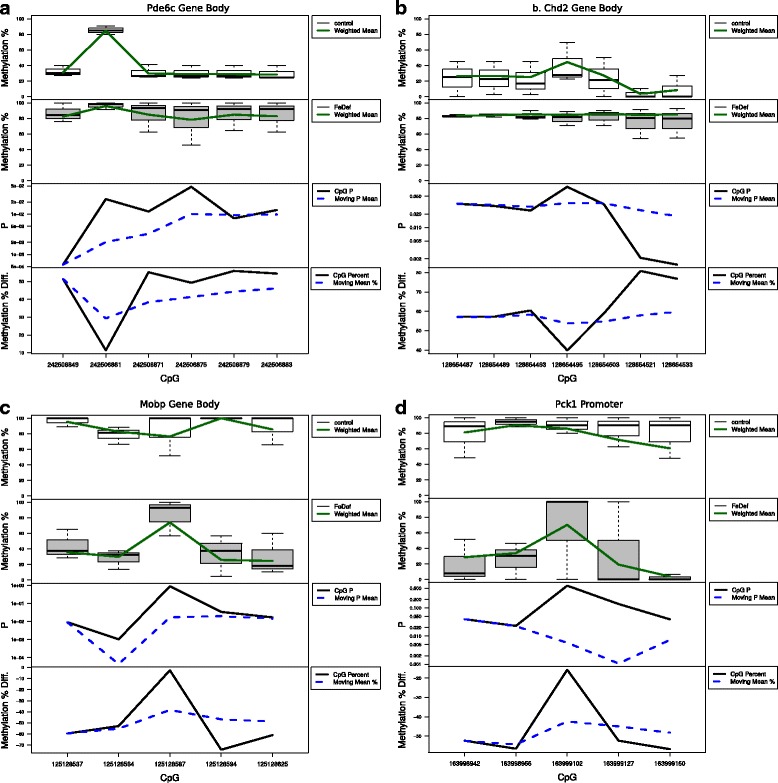
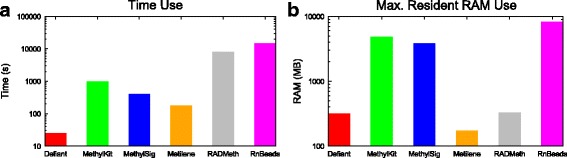
Results: We developed a new approach called Defiant to identify DMRs. Employing Weighted Welch Expansion (WWE), Defiant showed superior performance to other predictors in the series of benchmarking tests on artificial and real data. Defiant was subsequently used to investigate DNA methylation changes in iron-deficient rat hippocampus. Defiant identified DMRs close to genes associated with neuronal development and plasticity, which were not identified by its competitor. Importantly, Defiant runs between 5 to 479 times faster than currently available software packages. Also, Defiant accepts 10 different input formats widely used for DNA methylation data.
Conclusions: Defiant effectively identifies DMRs for whole-genome bisulfite sequencing (WGBS), reduced-representation bisulfite sequencing (RRBS), Tet-assisted bisulfite sequencing (TAB-seq), and HpaII tiny fragment enrichment by ligation-mediated PCR-tag (HELP) assays.
Keywords: Bisulfite sequencing; DNA Methylation; Differentially Methylated regions (DMR); Epigenetics; RRBS; WGBS.
Conflict of interest statement
Ethics approval and consent to participate
All experimental procedures involving the use of live animals were approved by the Institutional Animal Care and Use Committee review boards of the University of Minnesota, the Children’s Hospital of Philadelphia, and the University of Pennsylvania.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
Similar articles
-
HOME: a histogram based machine learning approach for effective identification of differentially methylated regions.BMC Bioinformatics. 2019 May 16;20(1):253. doi: 10.1186/s12859-019-2845-y. BMC Bioinformatics. 2019. PMID: 31096906 Free PMC article.
-
Sex-specific DNA methylation signatures of autism spectrum disorder from whole genome bisulfite sequencing of newborn blood.Biol Sex Differ. 2025 Apr 30;16(1):30. doi: 10.1186/s13293-025-00712-9. Biol Sex Differ. 2025. PMID: 40307894 Free PMC article.
-
Whole genome bisulfite sequencing of Down syndrome brain reveals regional DNA hypermethylation and novel disorder insights.Epigenetics. 2019 Jul;14(7):672-684. doi: 10.1080/15592294.2019.1609867. Epub 2019 May 6. Epigenetics. 2019. PMID: 31010359 Free PMC article.
-
Methodological aspects of whole-genome bisulfite sequencing analysis.Brief Bioinform. 2015 May;16(3):369-79. doi: 10.1093/bib/bbu016. Epub 2014 May 27. Brief Bioinform. 2015. PMID: 24867940 Review.
-
Methods for identifying differentially methylated regions for sequence- and array-based data.Brief Funct Genomics. 2016 Nov;15(6):485-490. doi: 10.1093/bfgp/elw018. Epub 2016 Jun 20. Brief Funct Genomics. 2016. PMID: 27323952 Review.
Cited by
-
Epigenetic control of ataxin-1 in multiple sclerosis.Ann Clin Transl Neurol. 2022 Aug;9(8):1186-1194. doi: 10.1002/acn3.51618. Epub 2022 Jul 28. Ann Clin Transl Neurol. 2022. PMID: 35903875 Free PMC article.
-
Identification of Novel Regulatory Regions Induced by Intrauterine Growth Restriction in Rat Islets.Endocrinology. 2022 Feb 1;163(2):bqab251. doi: 10.1210/endocr/bqab251. Endocrinology. 2022. PMID: 34894232 Free PMC article.
-
Nutritional influences on brain development.Acta Paediatr. 2018 Aug;107(8):1310-1321. doi: 10.1111/apa.14287. Epub 2018 Mar 22. Acta Paediatr. 2018. PMID: 29468731 Free PMC article. Review.
-
Exposure to Gestational Diabetes Enriches Immune-Related Pathways in the Transcriptome and Methylome of Human Amniocytes.J Clin Endocrinol Metab. 2020 Oct 1;105(10):3250-64. doi: 10.1210/clinem/dgaa466. J Clin Endocrinol Metab. 2020. PMID: 32687192 Free PMC article.
-
Analysis and Performance Assessment of the Whole Genome Bisulfite Sequencing Data Workflow: Currently Available Tools and a Practical Guide to Advance DNA Methylation Studies.Small Methods. 2022 Mar;6(3):e2101251. doi: 10.1002/smtd.202101251. Epub 2022 Jan 22. Small Methods. 2022. PMID: 35064762 Free PMC article. Review.
References
-
- Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, Scholer A, Wirbelauer C, Oakeley EJ, Gaidatzis D, Tiwari VK, Schubeler D. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature. 2011;480:490–495. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
