

Sialylation of EGFR by the ST6Gal-I sialyltransferase promotes EGFR activation and resistance to gefitinib-mediated cell death
- PMID: 29402301
- PMCID: PMC5800010
- DOI: 10.1186/s13048-018-0385-0
Sialylation of EGFR by the ST6Gal-I sialyltransferase promotes EGFR activation and resistance to gefitinib-mediated cell death
Abstract
Background: The ST6Gal-I sialyltransferase is upregulated in numerous cancers, and high expression of this enzyme correlates with poor patient prognosis in various malignancies, including ovarian cancer. Through its sialylation of a select cohort of cell surface receptors, ST6Gal-I modulates cell signaling to promote tumor cell survival. The goal of the present study was to investigate the influence of ST6Gal-I on another important receptor that controls cancer cell behavior, EGFR. Additionally, the effect of ST6Gal-I on cancer cells treated with the common EGFR inhibitor, gefitinib, was evaluated.
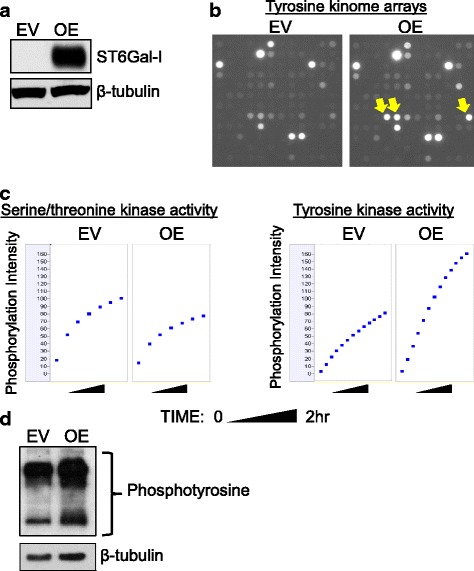
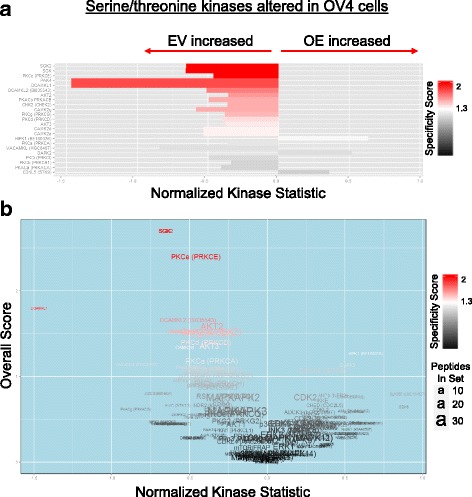
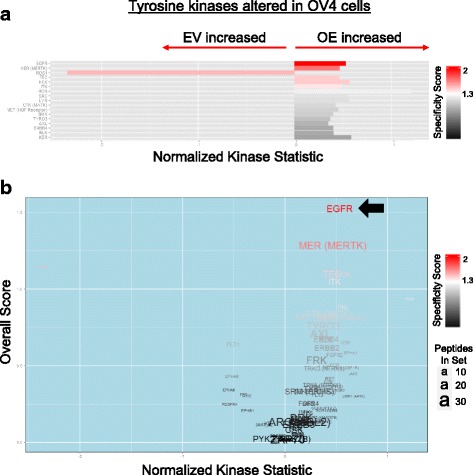
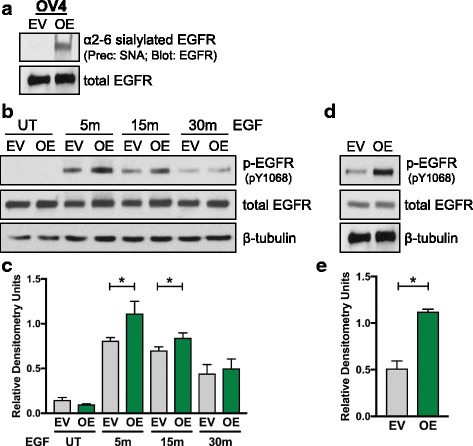
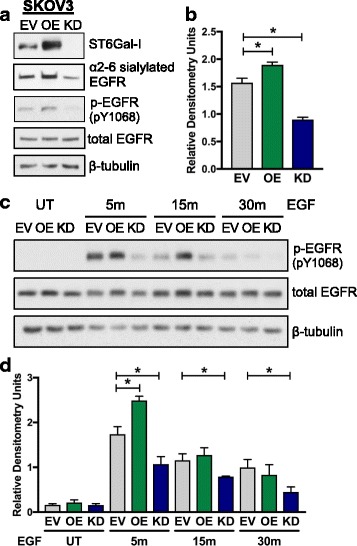
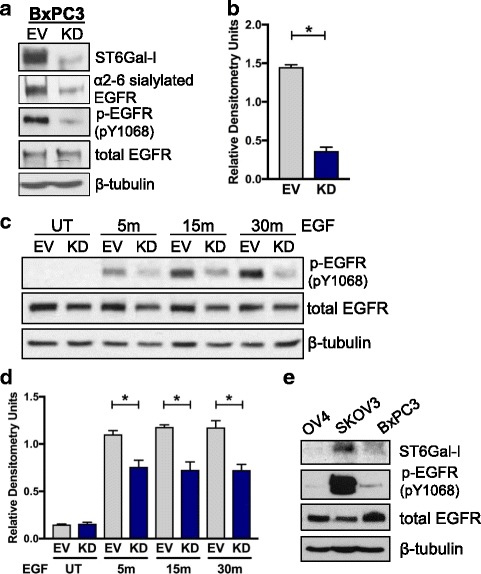
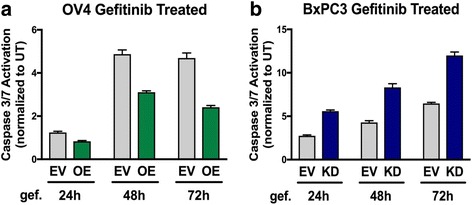
Results: Using the OV4 ovarian cancer cell line, which lacks endogenous ST6Gal-I expression, a kinomics assay revealed that cells with forced overexpression of ST6Gal-I exhibited increased global tyrosine kinase activity, a finding confirmed by immunoblotting whole cell lysates with an anti-phosphotyrosine antibody. Interestingly, the kinomics assay suggested that one of the most highly activated tyrosine kinases in ST6Gal-I-overexpressing OV4 cells was EGFR. Based on these findings, additional analyses were performed to investigate the effect of ST6Gal-I on EGFR activation. To this end, we utilized, in addition to OV4 cells, the SKOV3 ovarian cancer cell line, engineered with both ST6Gal-I overexpression and knockdown, as well as the BxPC3 pancreatic cancer cell line with knockdown of ST6Gal-I. In all three cell lines, we determined that EGFR is a substrate of ST6Gal-I, and that the sialylation status of EGFR directly correlates with ST6Gal-I expression. Cells with differential ST6Gal-I expression were subsequently evaluated for EGFR tyrosine phosphorylation. Cells with high ST6Gal-I expression were found to have elevated levels of basal and EGF-induced EGFR activation. Conversely, knockdown of ST6Gal-I greatly attenuated EGFR activation, both basally and post EGF treatment. Finally, to illustrate the functional importance of ST6Gal-I in regulating EGFR-dependent survival, cells were treated with gefitinib, an EGFR inhibitor widely used for cancer therapy. These studies showed that ST6Gal-I promotes resistance to gefitinib-mediated apoptosis, as measured by caspase activity assays.
Conclusion: Results herein indicate that ST6Gal-I promotes EGFR activation and protects against gefitinib-mediated cell death. Establishing the tumor-associated ST6Gal-I sialyltransferase as a regulator of EGFR provides novel insight into the role of glycosylation in growth factor signaling and chemoresistance.
Keywords: Epidermal growth factor receptor (EGFR) cell signaling; Gefitinib; Glycosylation; Kinomics; Tumor cell biology; Tyrosine kinase; β-galactoside α2-6 sialyltransferase 1 (ST6GAL1).
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Swindall AF, Londono-Joshi AI, Schultz MJ, Fineberg N, Buchsbaum DJ, Bellis SL. ST6Gal-I protein expression is upregulated in human epithelial tumors and correlates with stem cell markers in normal tissues and colon cancer cell lines. Cancer Res. 2013;73(7):2368–2378. doi: 10.1158/0008-5472.CAN-12-3424. - DOI - PMC - PubMed
-
- Schultz MJ, Holdbrooks AT, Chakraborty A, Grizzle WE, Landen CN, Buchsbaum DJ, et al. The tumor-associated glycosyltransferase ST6Gal-I regulates stem cell transcription factors and confers a cancer stem cell phenotype. Cancer Res. 2016;76(13):3978–3988. doi: 10.1158/0008-5472.CAN-15-2834. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
