

Imbalance between firing homeostasis and synaptic plasticity drives early-phase Alzheimer's disease
- PMID: 29403035
- PMCID: PMC6533171
- DOI: 10.1038/s41593-018-0080-x
Imbalance between firing homeostasis and synaptic plasticity drives early-phase Alzheimer's disease
Abstract
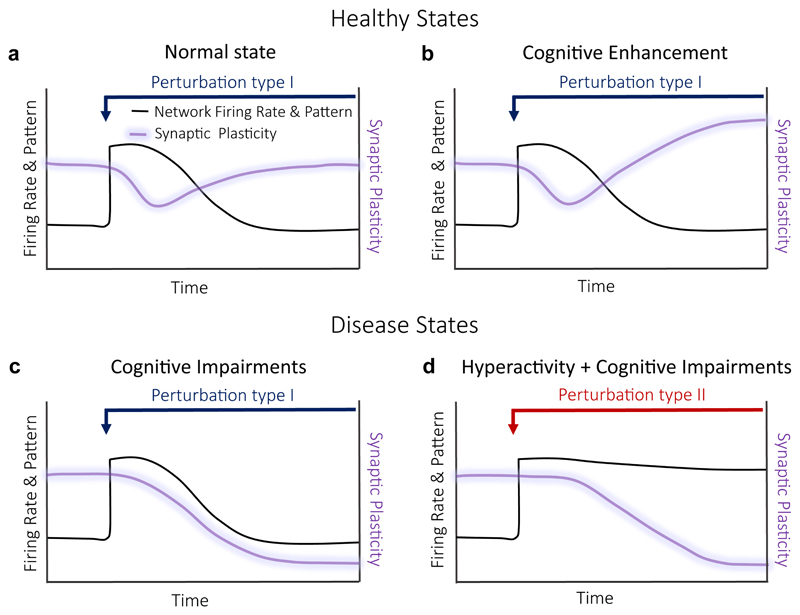
During recent years, the preclinical stage of Alzheimer's disease (AD) has become a major focus of research. Continued failures in clinical trials and the realization that early intervention may offer better therapeutic outcome triggered a conceptual shift from late-stage AD pathology to early-stage pathophysiology. While much effort has been directed at understanding the factors initiating AD, little is known about the principle basis underlying the disease progression at its early stages. In this Perspective, we suggest a hypothesis to explain the transition from 'silent' signatures of aberrant neural circuit activity to clinically evident memory impairments. Namely, we propose that failures in firing homeostasis and imbalance between firing stability and synaptic plasticity in cortico-hippocampal circuits represent the driving force of early disease progression. We analyze the main types of possible homeostatic failures and provide the essential conceptual framework for examining the causal link between dysregulation of firing homeostasis, aberrant neural circuit activity and memory-related plasticity impairments associated with early AD.
Figures
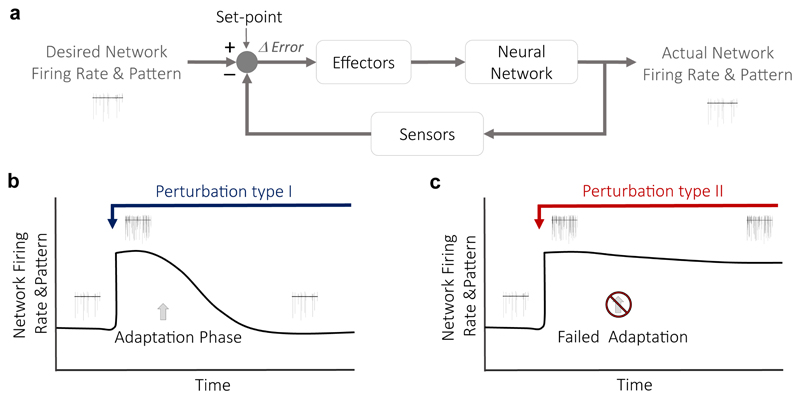
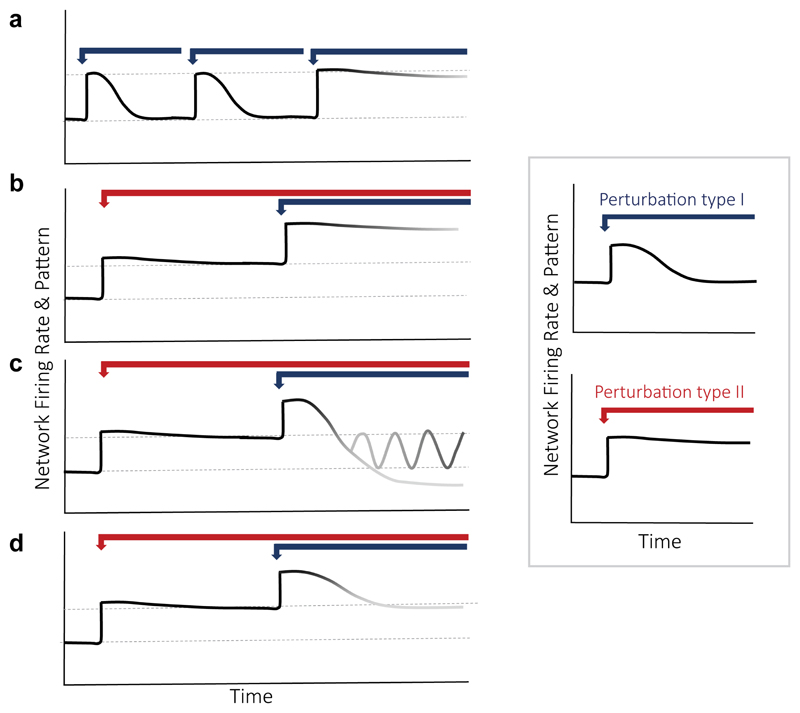
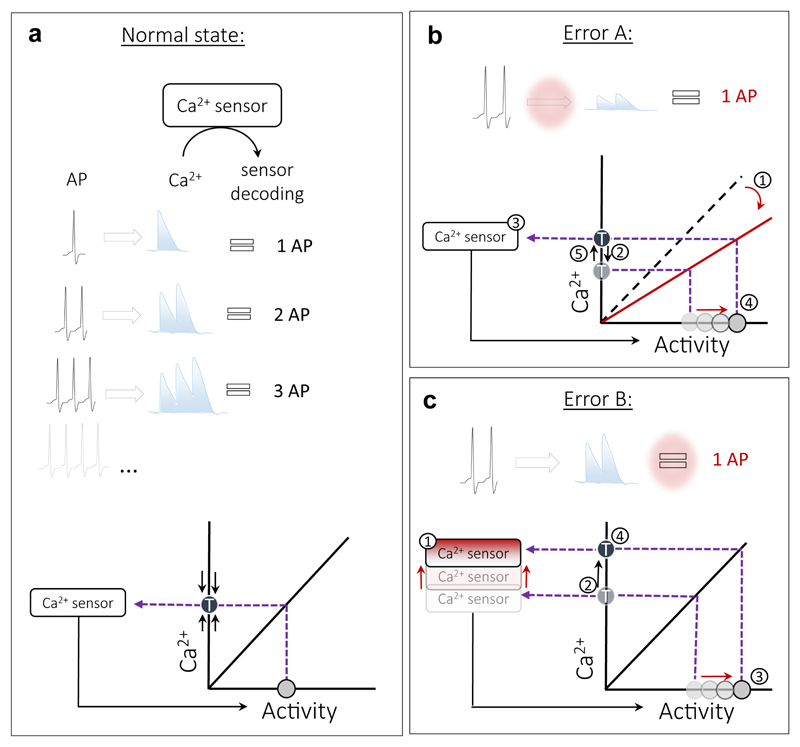
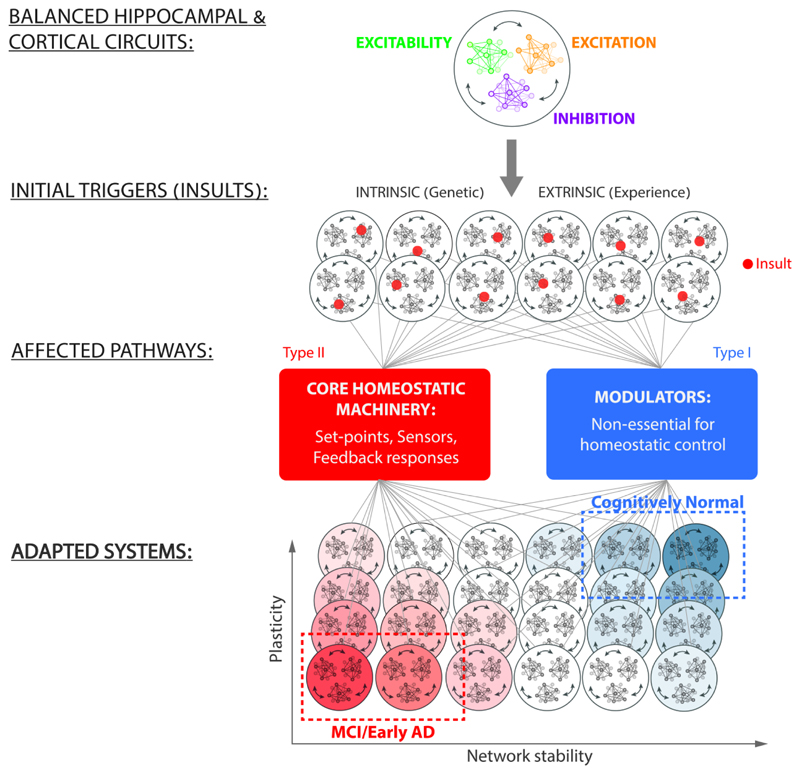
References
-
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. - PubMed
-
- Herrup K. The case for rejecting the amyloid cascade hypothesis. Nat Neurosci. 2015:794–799. - PubMed
-
- De Strooper B, Karran E. The Cellular Phase of Alzheimer’s Disease. Cell. 2016;164:603–615. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
