

The mTOR-S6K pathway links growth signalling to DNA damage response by targeting RNF168
- PMID: 29403037
- PMCID: PMC5826806
- DOI: 10.1038/s41556-017-0033-8
The mTOR-S6K pathway links growth signalling to DNA damage response by targeting RNF168
Abstract
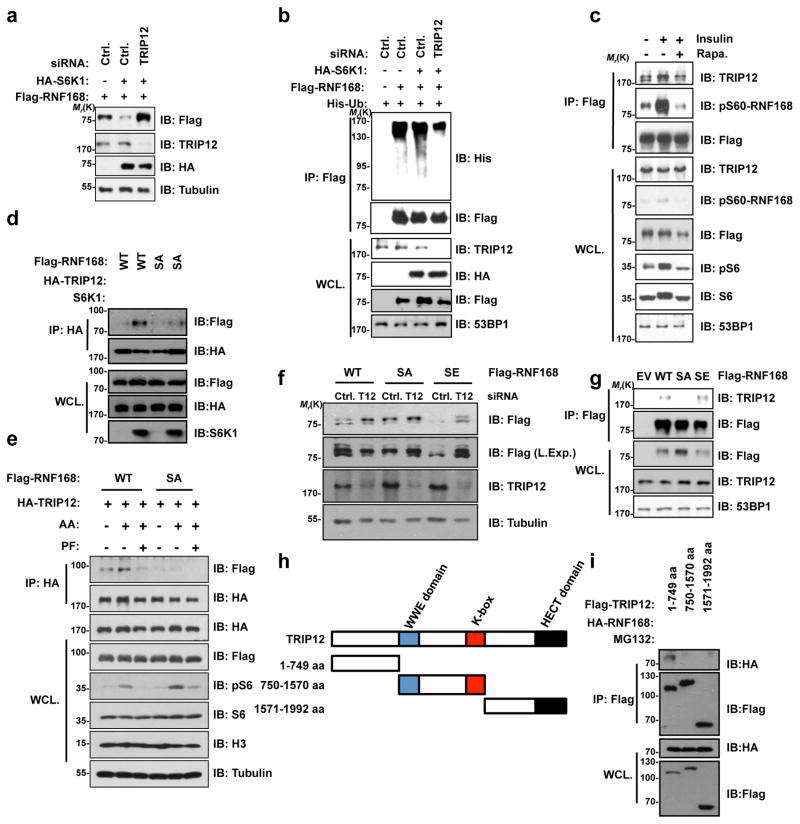
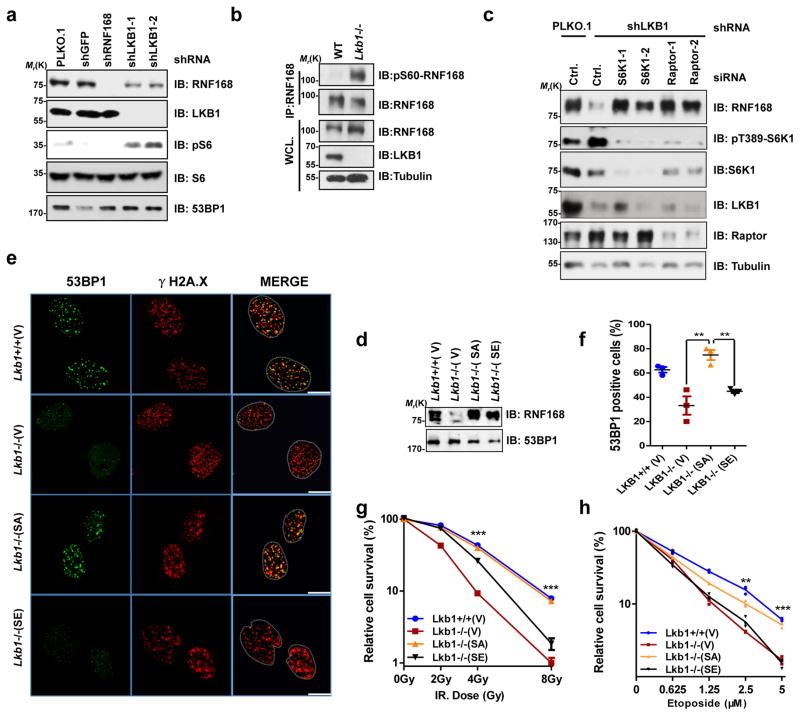
Growth signals, such as extracellular nutrients and growth factors, have substantial effects on genome integrity; however, the direct underlying link remains unclear. Here, we show that the mechanistic target of rapamycin (mTOR)-ribosomal S6 kinase (S6K) pathway, a central regulator of growth signalling, phosphorylates RNF168 at Ser60 to inhibit its E3 ligase activity, accelerate its proteolysis and impair its function in the DNA damage response, leading to accumulated unrepaired DNA and genome instability. Moreover, loss of the tumour suppressor liver kinase B1 (LKB1; also known as STK11) hyperactivates mTOR complex 1 (mTORC1)-S6K signalling and decreases RNF168 expression, resulting in defects in the DNA damage response. Expression of a phospho-deficient RNF168-S60A mutant rescues the DNA damage repair defects and suppresses tumorigenesis caused by Lkb1 loss. These results reveal an important function of mTORC1-S6K signalling in the DNA damage response and suggest a general mechanism that connects cell growth signalling to genome stability control.
Conflict of interest statement
The authors declare no competing financial interests.
Figures





References
-
- Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annual review of genetics. 2011;45:247–271. - PubMed
-
- Khanna KK, Jackson SP. DNA double-strand breaks: signaling, repair and the cancer connection. Nature genetics. 2001;27:247–254. - PubMed
-
- Jackson SP. Sensing and repairing DNA double-strand breaks. Carcinogenesis. 2002;23:687–696. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
