

Helicobacter pylori and Gastric Cancer: Adaptive Cellular Mechanisms Involved in Disease Progression
- PMID: 29403459
- PMCID: PMC5786524
- DOI: 10.3389/fmicb.2018.00005
Helicobacter pylori and Gastric Cancer: Adaptive Cellular Mechanisms Involved in Disease Progression
Abstract
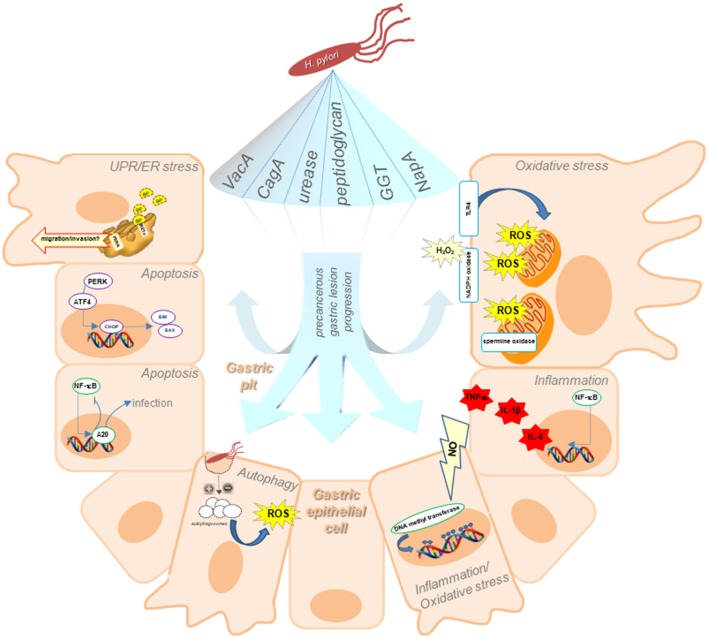
Helicobacter pylori (H. pylori) infection is the major risk factor associated with the development of gastric cancer. The transition from normal mucosa to non-atrophic gastritis, triggered primarily by H. pylori infection, initiates precancerous lesions which may then progress to atrophic gastritis and intestinal metaplasia. Further progression to dysplasia and gastric cancer is generally believed to be attributable to processes that no longer require the presence of H. pylori. The responses that develop upon H. pylori infection are directly mediated through the action of bacterial virulence factors, which drive the initial events associated with transformation of infected gastric cells. Besides genetic and to date poorly defined environmental factors, alterations in gastric cell stress-adaptive mechanisms due to H. pylori appear to be crucial during chronic infection and gastric disease progression. Firstly, H. pylori infection promotes gastric cell death and reduced epithelial cell turnover in the majority of infected cells, resulting in primary tissue lesions associated with an initial inflammatory response. However, in the remaining gastric cell population, adaptive responses are induced that increase cell survival and proliferation, resulting in the acquisition of potentially malignant characteristics that may lead to precancerous gastric lesions. Thus, deregulation of these intrinsic survival-related responses to H. pylori infection emerge as potential culprits in promoting disease progression. This review will highlight the most relevant cellular adaptive mechanisms triggered upon H. pylori infection, including endoplasmic reticulum stress and the unfolded protein response, autophagy, oxidative stress, and inflammation, together with a subsequent discussion on how these factors may participate in the progression of a precancerous lesion. Finally, this review will shed light on how these mechanisms may be exploited as pharmacological targets, in the perspective of opening up new therapeutic alternatives for non-invasive risk control in gastric cancer.
Keywords: Helicobacter pylori; autophagy; endoplasmic reticulum stress; gastric cancer; inflammation; oxidative stress; precancerous lesion.
Figures
References
-
- Axten J. M., Medina J. R., Feng Y., Shu A., Romeril S. P., Grant S. W., et al. (2012). Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-p yrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J. Med. Chem. 55, 7193–7207. 10.1021/jm300713s - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources