

The influence of phylodynamic model specifications on parameter estimates of the Zika virus epidemic
- PMID: 29403651
- PMCID: PMC5789282
- DOI: 10.1093/ve/vex044
The influence of phylodynamic model specifications on parameter estimates of the Zika virus epidemic
Abstract
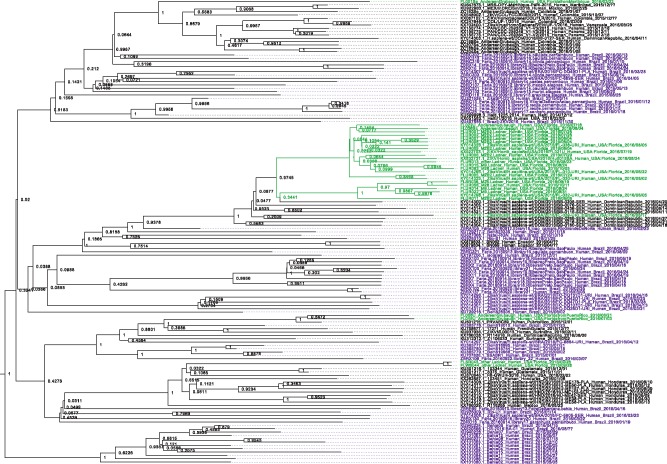
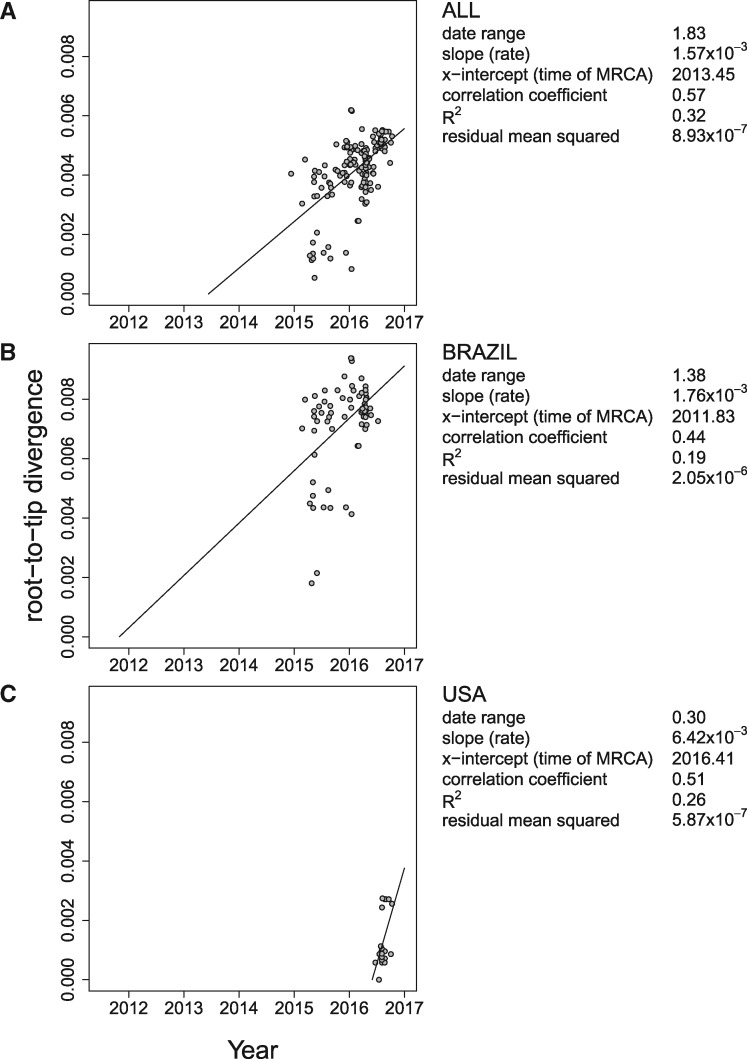
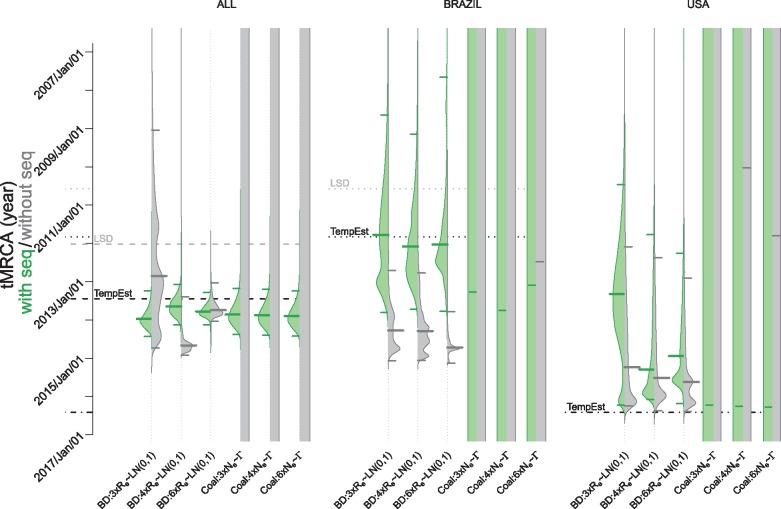
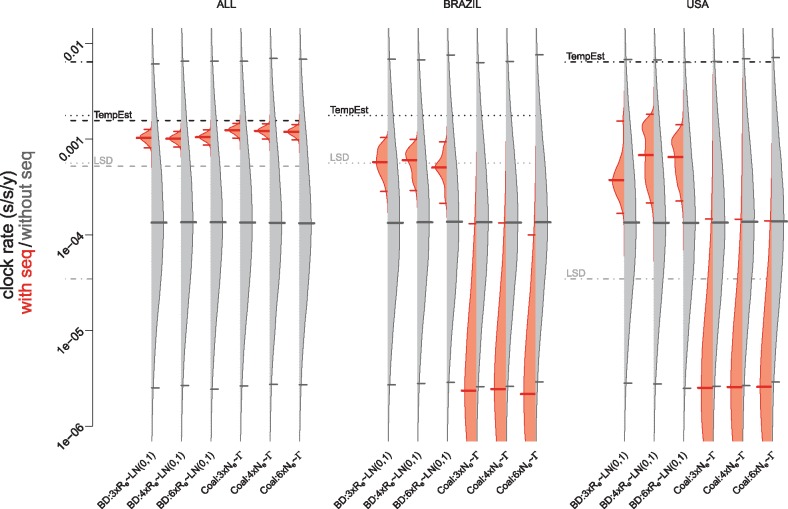
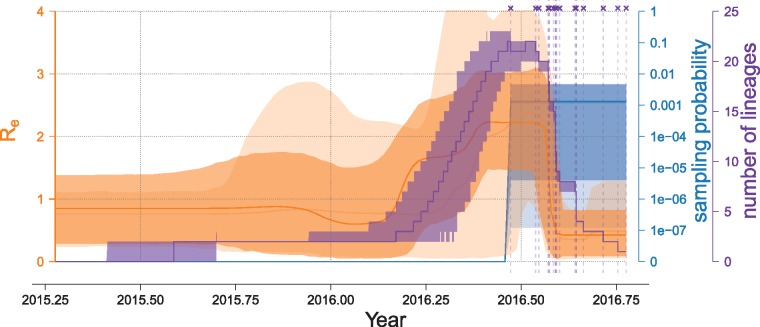
Each new virus introduced into the human population could potentially spread and cause a worldwide epidemic. Thus, early quantification of epidemic spread is crucial. Real-time sequencing followed by Bayesian phylodynamic analysis has proven to be extremely informative in this respect. Bayesian phylodynamic analyses require a model to be chosen and prior distributions on model parameters to be specified. We study here how choices regarding the tree prior influence quantification of epidemic spread in an emerging epidemic by focusing on estimates of the parameters clock rate, tree height, and reproductive number in the currently ongoing Zika virus epidemic in the Americas. While parameter estimates are quite robust to reasonable variations in the model settings when studying the complete data set, it is impossible to obtain unequivocal estimates when reducing the data to local Zika epidemics in Brazil and Florida, USA. Beyond the empirical insights, this study highlights the conceptual differences between the so-called birth-death and coalescent tree priors: while sequence sampling times alone can strongly inform the tree height and reproductive number under a birth-death model, the coalescent tree height prior is typically only slightly influenced by this information. Such conceptual differences together with non-trivial interactions of different priors complicate proper interpretation of empirical results. Overall, our findings indicate that phylodynamic analyses of early viral spread data must be carried out with care as data sets may not necessarily be informative enough yet to provide estimates robust to prior settings. It is necessary to do a robustness check of these data sets by scanning several models and prior distributions. Only if the posterior distributions are robust to reasonable changes of the prior distribution, the parameter estimates can be trusted. Such robustness tests will help making real-time phylodynamic analyses of spreading epidemic more reliable in the future.
Keywords: molecular epidemiology; start of epidemic; substitution rate; tree height; tree prior.
Figures
References
-
- Anderson R. M., May R. M. (1991) Infectious Diseases of Humans: Dynamics and Control. Oxford: Oxford University Press.
-
- Drummond A. J. et al. (2005) ‘Bayesian Coalescent Inference of past Population Dynamics from Molecular Sequences’, Molecular Biology and Evolution, 22: 1185–92. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
