

Splenic macrophages are required for protective innate immunity against West Nile virus
- PMID: 29408905
- PMCID: PMC5800658
- DOI: 10.1371/journal.pone.0191690
Splenic macrophages are required for protective innate immunity against West Nile virus
Abstract
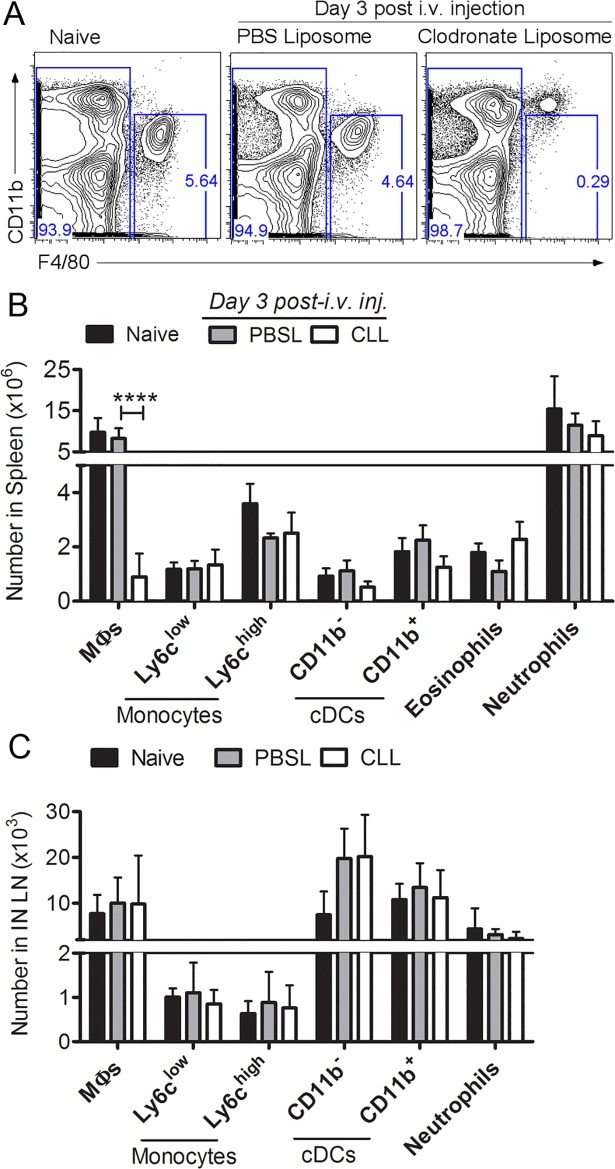
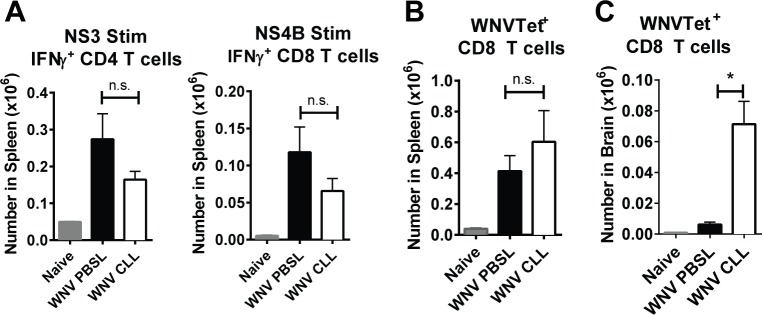
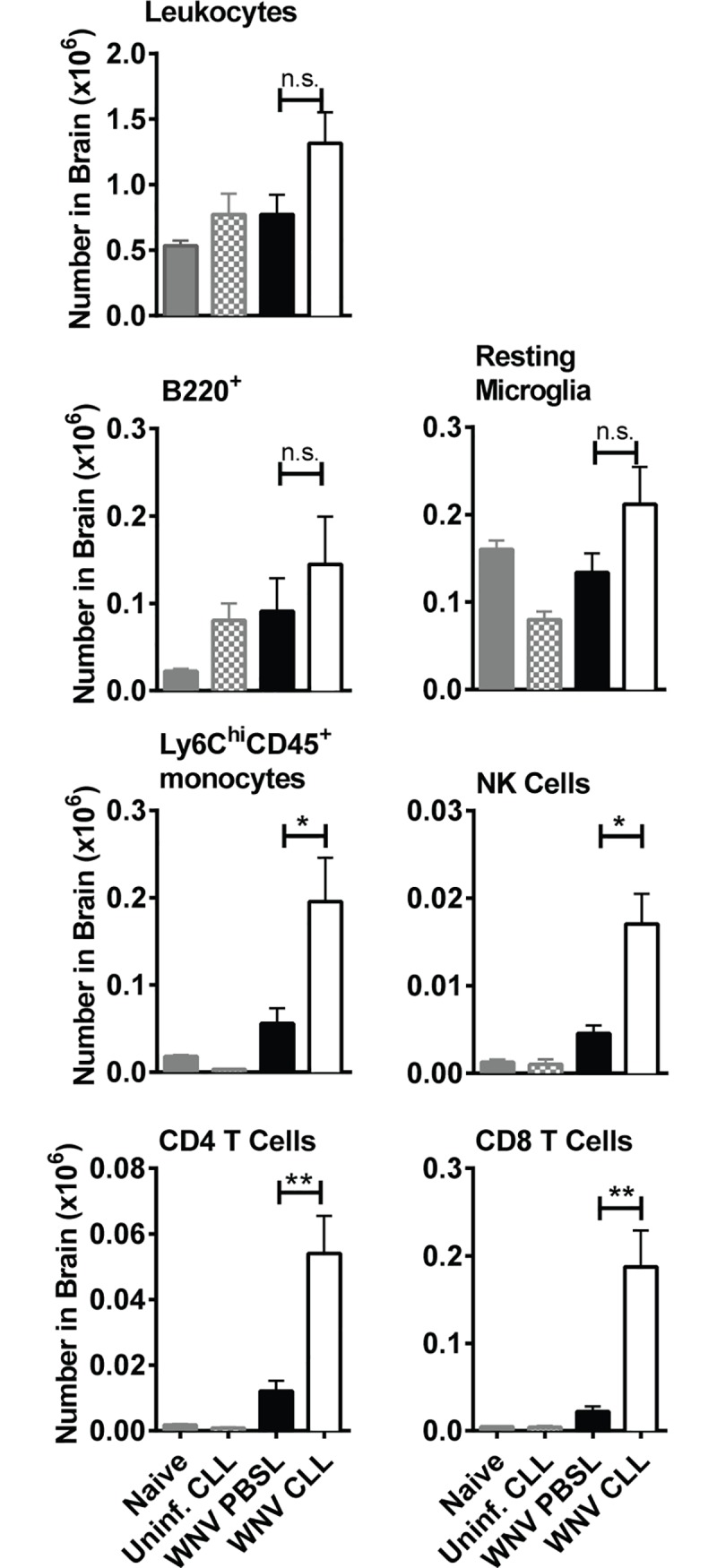
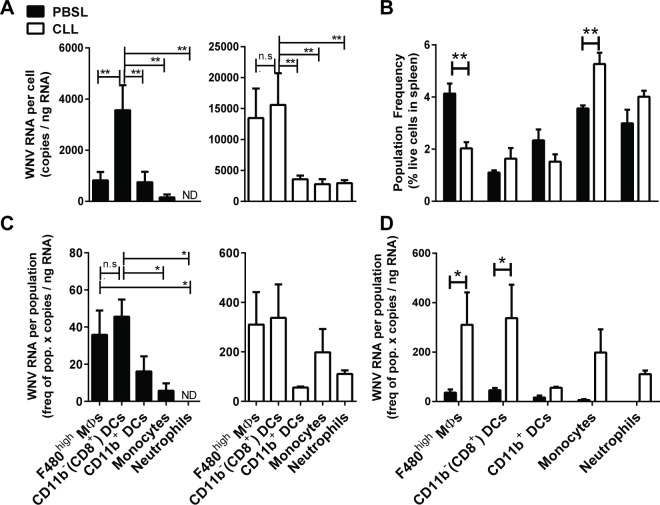
Although the spleen is a major site for West Nile virus (WNV) replication and spread, relatively little is known about which innate cells in the spleen replicate WNV, control viral dissemination, and/or prime innate and adaptive immune responses. Here we tested if splenic macrophages (MΦs) were necessary for control of WNV infection. We selectively depleted splenic MΦs, but not draining lymph node MΦs, by injecting mice intravenously with clodronate liposomes several days prior to infecting them with WNV. Mice missing splenic MΦs succumbed to WNV infection after an increased and accelerated spread of virus to the spleen and the brain. WNV-specific Ab and CTL responses were normal in splenic MΦ-depleted mice; however, numbers of NK cells and CD4 and CD8 T cells were significantly increased in the brains of infected mice. Splenic MΦ deficiency led to increased WNV in other splenic innate immune cells including CD11b- DCs, newly formed MΦs and monocytes. Unlike other splenic myeloid subsets, splenic MΦs express high levels of mRNAs encoding the complement protein C1q, the apoptotic cell clearance protein Mertk, the IL-18 cytokine and the FcγR1 receptor. Splenic MΦ-deficient mice may be highly susceptible to WNV infection in part to a deficiency in C1q, Mertk, IL-18 or Caspase 12 expression.
Conflict of interest statement
Figures




References
-
- Petersen L. R., Brault A. C., and Nasci R. S. 2013. West Nile virus: review of the literature. JAMA. 310:308–15. doi: 10.1001/jama.2013.8042 - DOI - PMC - PubMed
-
- Roehrig J. T. 2013. West nile virus in the United States—a historical perspective. Viruses. 5:3088–108. doi: 10.3390/v5123088 - DOI - PMC - PubMed
-
- Murray K. O., Ruktanonchai D., Hesalroad D., Fonken E., and Nolan M. S. 2013. West Nile virus, Texas, USA, 2012. Emerg. Infect. Dis. 19:1836–8. doi: 10.3201/eid1911.130768 - DOI - PMC - PubMed
-
- Beasley D. W., Barrett A. D., and Tesh R. B. 2013. Resurgence of West Nile neurologic disease in the United States in 2012: what happened? What needs to be done? Antiviral Res. 99:1–5. doi: 10.1016/j.antiviral.2013.04.015 - DOI - PubMed
-
- Diamond M.S. 2009. Progress on the development of therapeutics against West Nile virus. Antiviral Res. 83:214–27. doi: 10.1016/j.antiviral.2009.05.006 - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
