

Integration of human pancreatic islet genomic data refines regulatory mechanisms at Type 2 Diabetes susceptibility loci
- PMID: 29412141
- PMCID: PMC5828664
- DOI: 10.7554/eLife.31977
Integration of human pancreatic islet genomic data refines regulatory mechanisms at Type 2 Diabetes susceptibility loci
Abstract
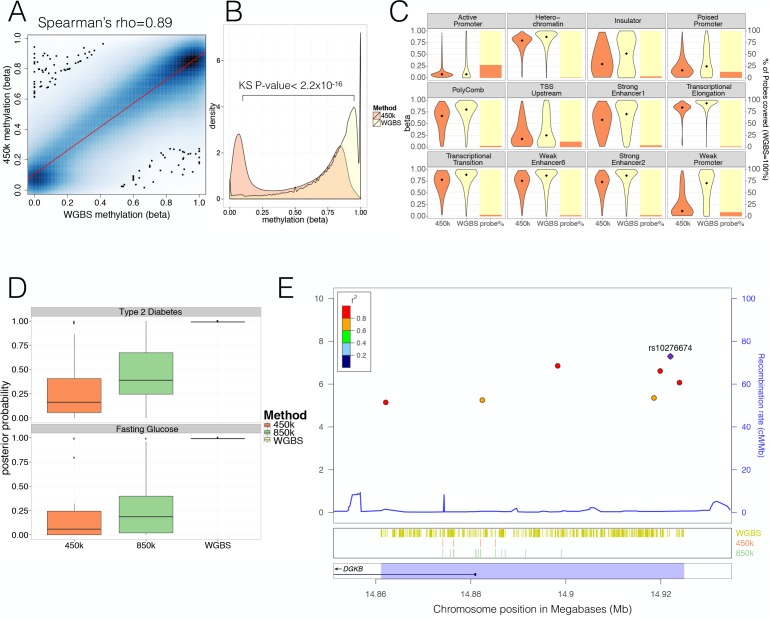
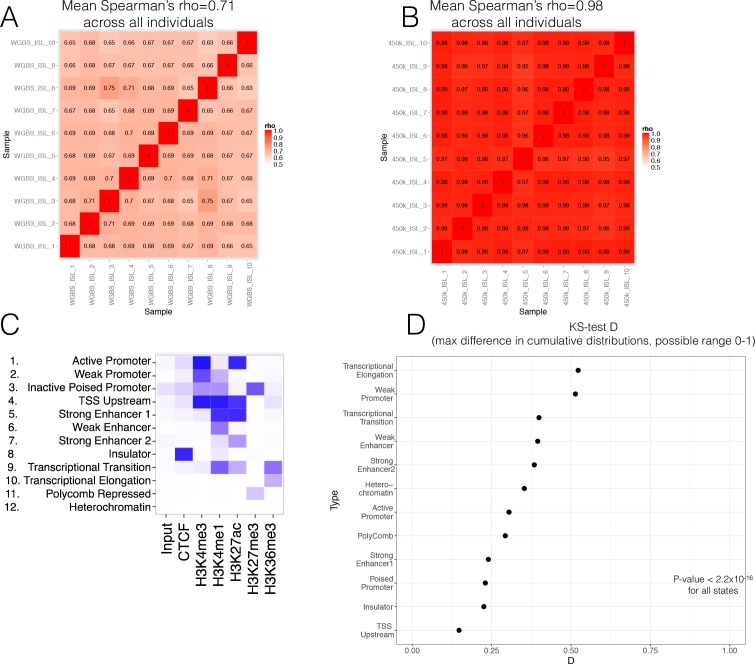
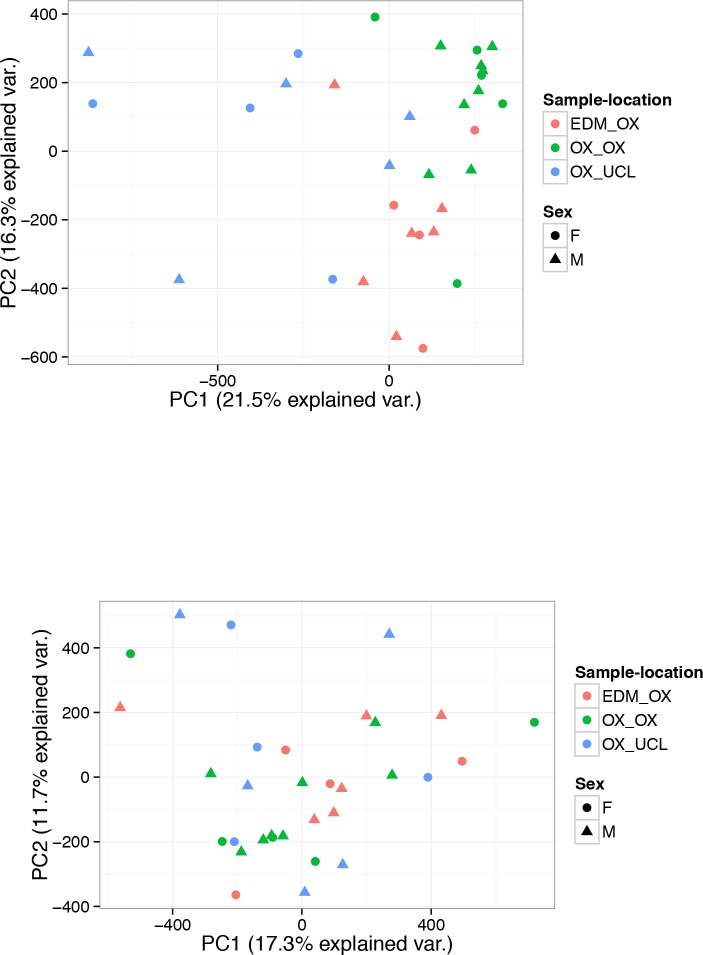
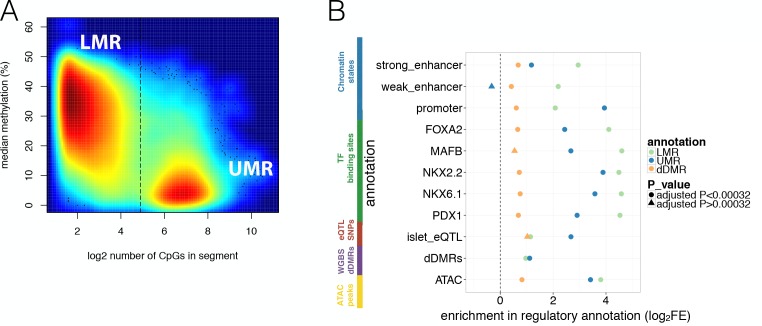
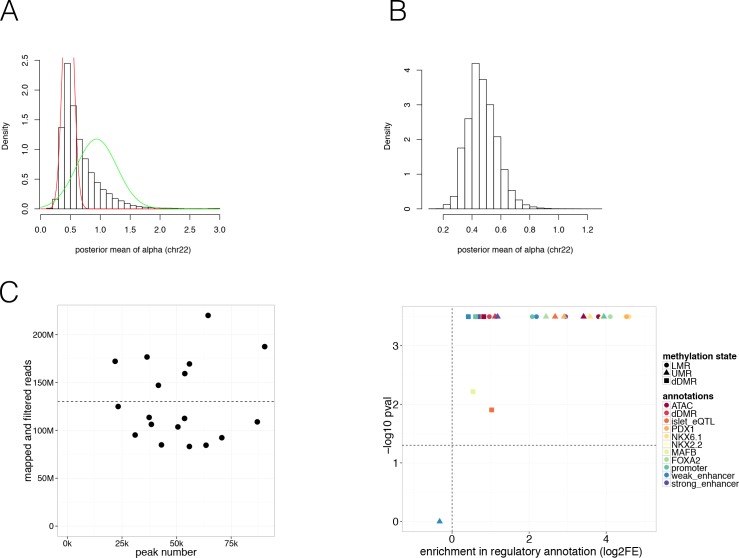
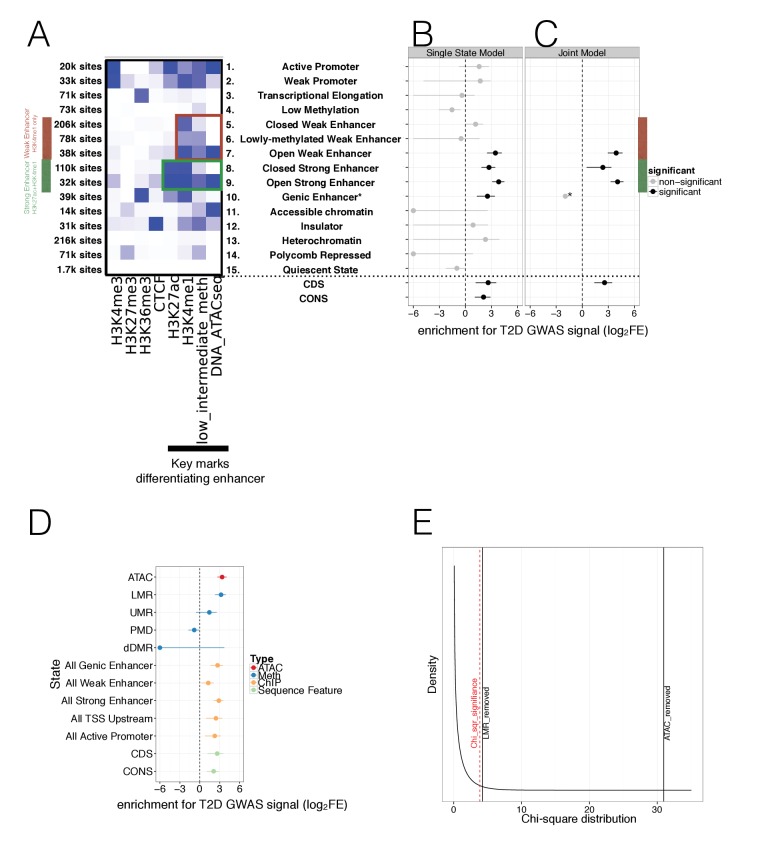
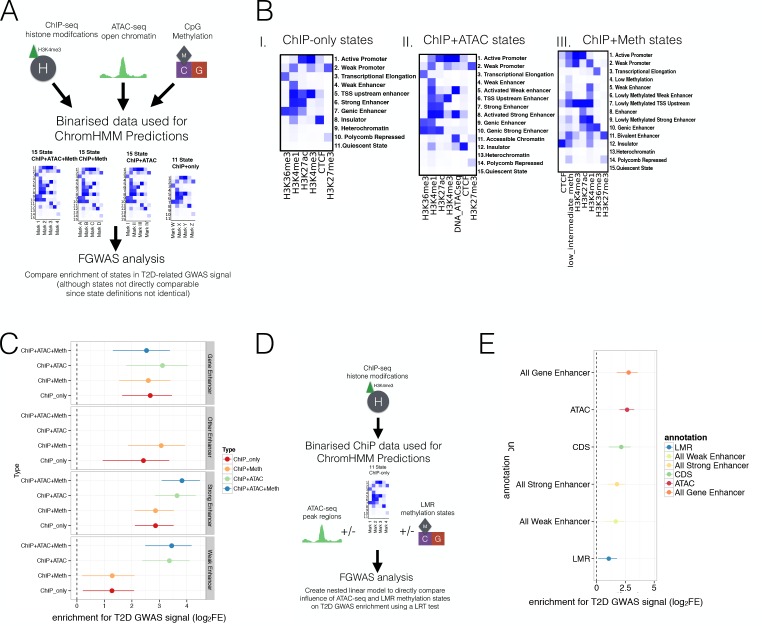
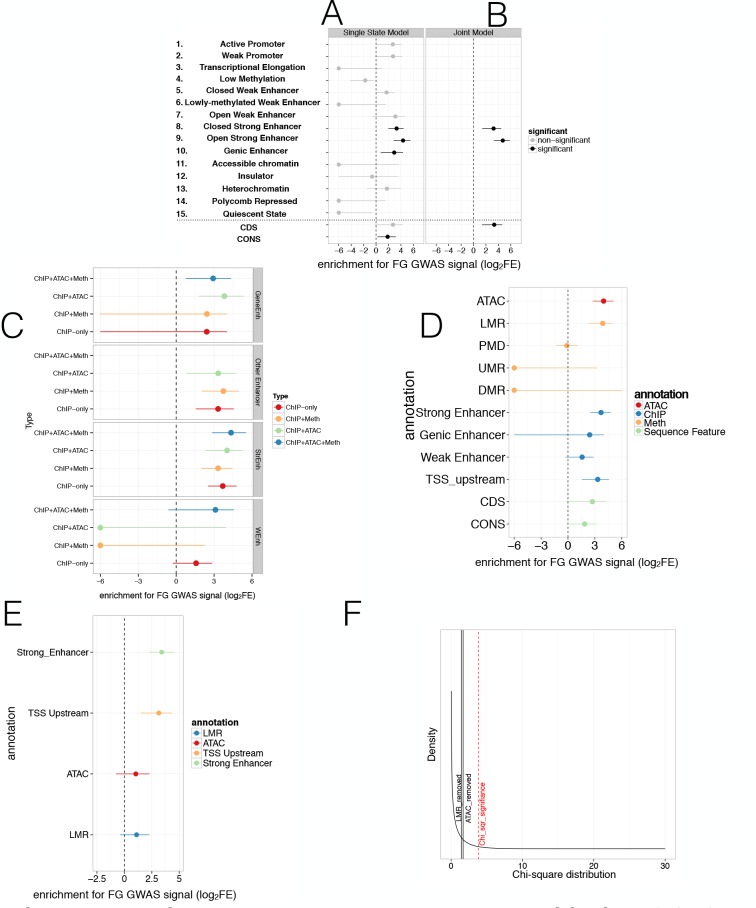
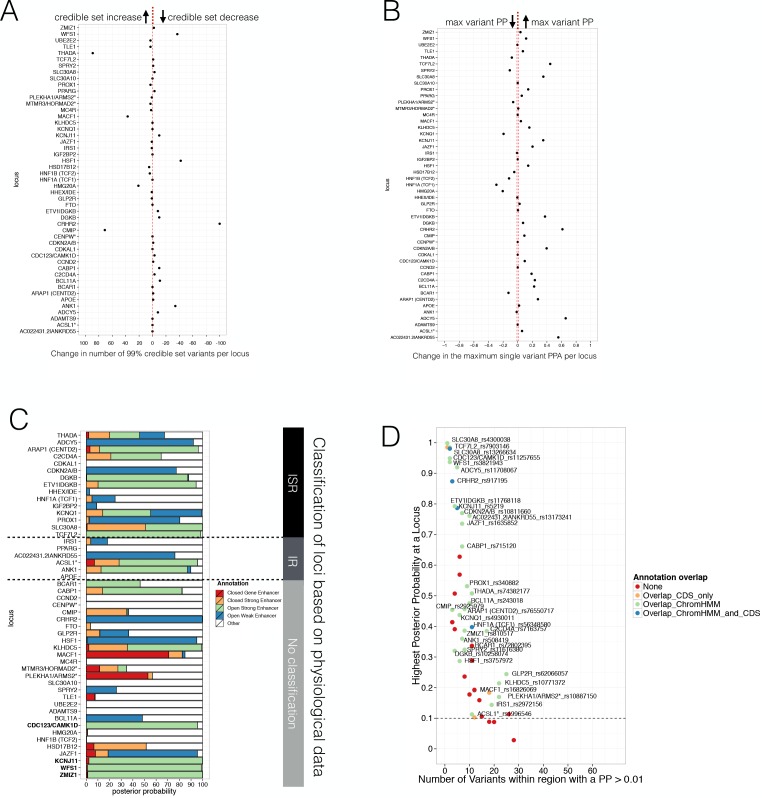
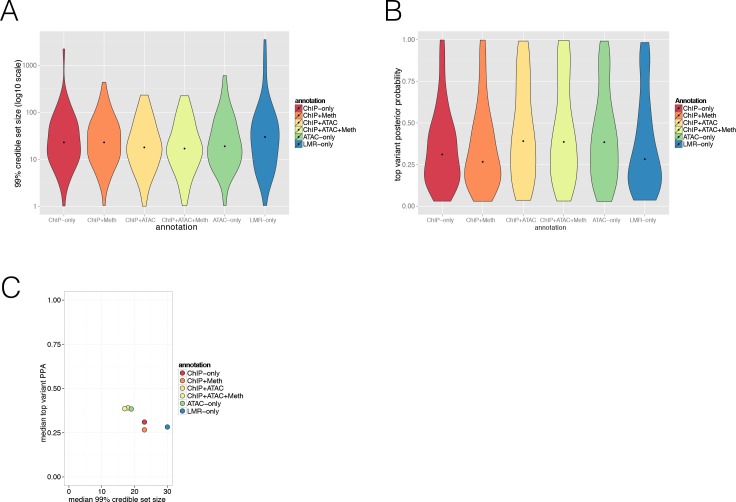
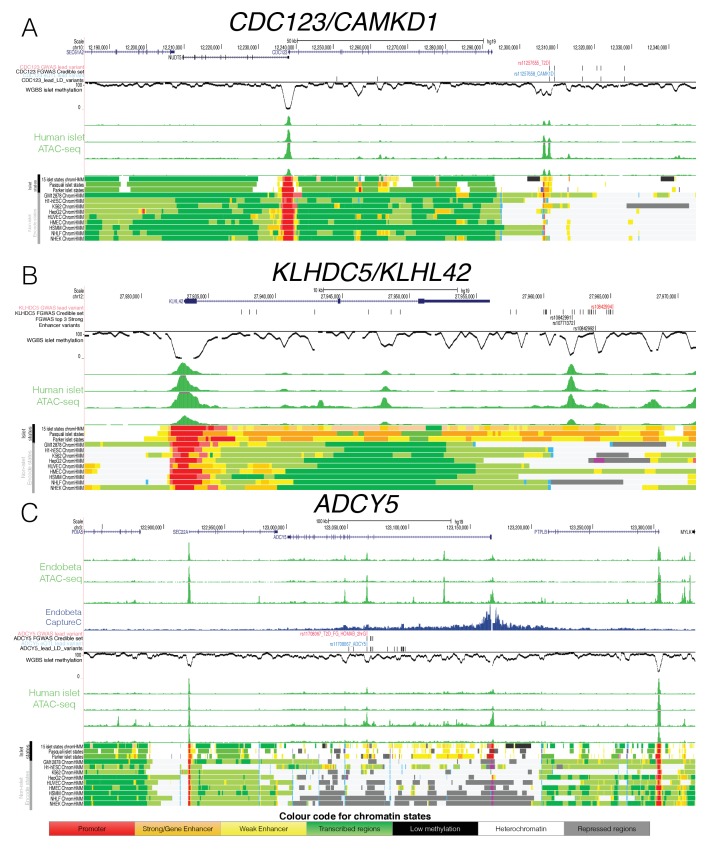
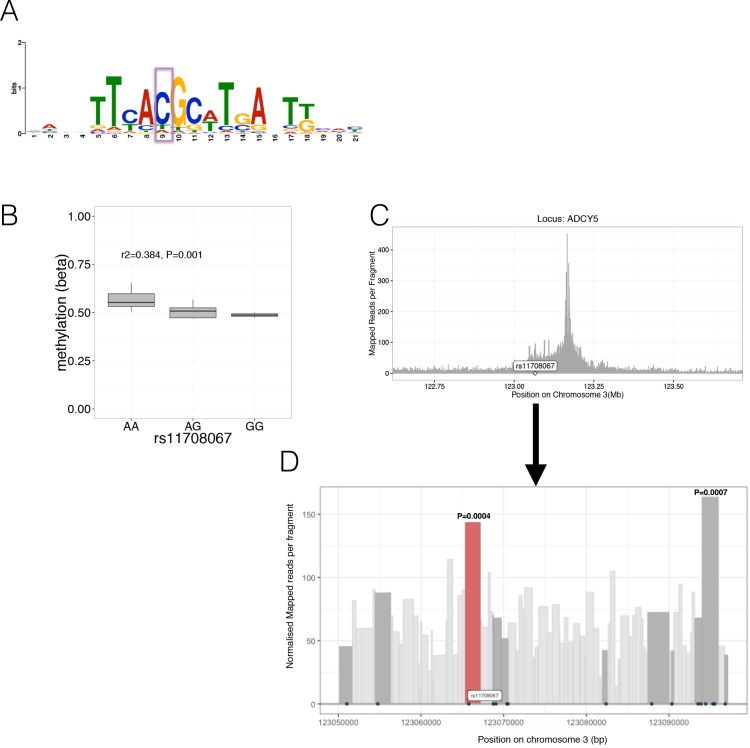
Human genetic studies have emphasised the dominant contribution of pancreatic islet dysfunction to development of Type 2 Diabetes (T2D). However, limited annotation of the islet epigenome has constrained efforts to define the molecular mechanisms mediating the, largely regulatory, signals revealed by Genome-Wide Association Studies (GWAS). We characterised patterns of chromatin accessibility (ATAC-seq, n = 17) and DNA methylation (whole-genome bisulphite sequencing, n = 10) in human islets, generating high-resolution chromatin state maps through integration with established ChIP-seq marks. We found enrichment of GWAS signals for T2D and fasting glucose was concentrated in subsets of islet enhancers characterised by open chromatin and hypomethylation, with the former annotation predominant. At several loci (including CDC123, ADCY5, KLHDC5) the combination of fine-mapping genetic data and chromatin state enrichment maps, supplemented by allelic imbalance in chromatin accessibility pinpointed likely causal variants. The combination of increasingly-precise genetic and islet epigenomic information accelerates definition of causal mechanisms implicated in T2D pathogenesis.
Keywords: GWAS; Type 2 Diabetes; epigenetics; evolutionary biology; genomics; human; human biology; human pancreatic islets; medicine.
© 2018, Thurner et al.
Conflict of interest statement
MT, Mv, JT, AM, VN, AB, KG, AB, CB, CB, RL, SB, VR, AG No competing interests declared, MM Senior editor, <italic>eLife</italic>
Figures
References
-
- Carlson M, Maintainer B. TxDb.Hsapiens.UCSC.hg19.knownGene: Annotation Package for TxDb Object(s) R package version 3.2.22015
Publication types
MeSH terms
Substances
Grants and funding
- U01 DK085545/DK/NIDDK NIH HHS/United States
- MR/L020149/1/MRC_/Medical Research Council/United Kingdom
- R01 DK098032/DK/NIDDK NIH HHS/United States
- 106130/WT_/Wellcome Trust/United Kingdom
- U01 DK105535/DK/NIDDK NIH HHS/United States
- 90532/WT_/Wellcome Trust/United Kingdom
- 90367/WT_/Wellcome Trust/United Kingdom
- R01 MH090941/MH/NIMH NIH HHS/United States
- 098381/WT_/Wellcome Trust/United Kingdom
- 200837/Z/16/Z/WT_/Wellcome Trust/United Kingdom
- R01-DK098032/DK/NIDDK NIH HHS/United States
- R01-MH090941/MH/NIMH NIH HHS/United States
- 98381/WT_/Wellcome Trust/United Kingdom
- 095101/Z/10/Z/WT_/Wellcome Trust/United Kingdom
- U01-DK085545/DK/NIDDK NIH HHS/United States
- 090367/WT_/Wellcome Trust/United Kingdom
- 090532/WT_/Wellcome Trust/United Kingdom
- 099673/Z/12/Z/WT_/Wellcome Trust/United Kingdom
- U01-DK105535/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
