

Peptide Level Turnover Measurements Enable the Study of Proteoform Dynamics
- PMID: 29414762
- PMCID: PMC5930408
- DOI: 10.1074/mcp.RA118.000583
Peptide Level Turnover Measurements Enable the Study of Proteoform Dynamics
Abstract
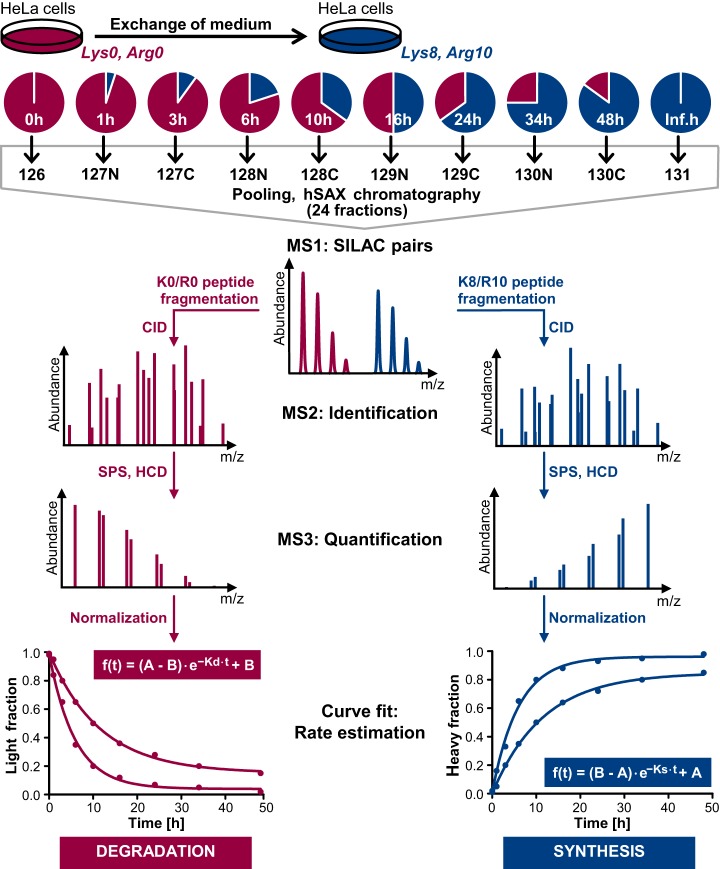
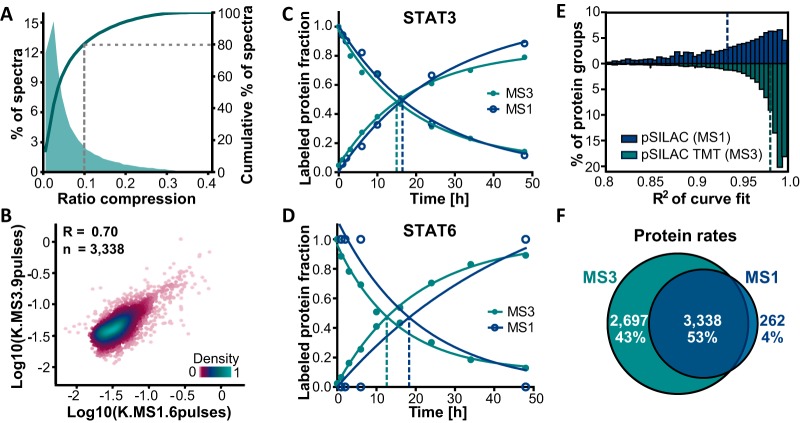
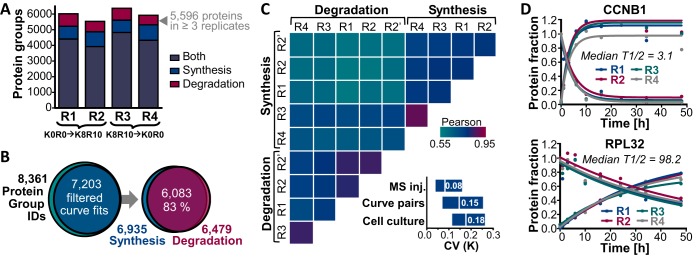
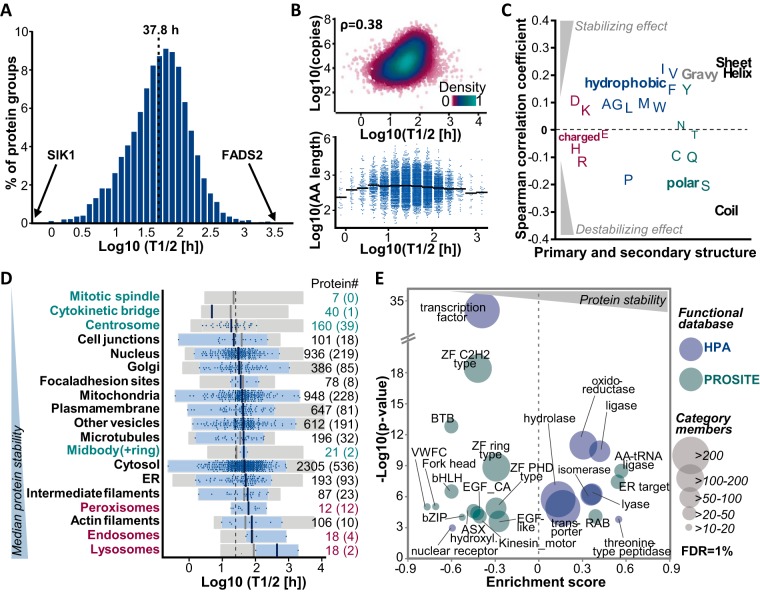
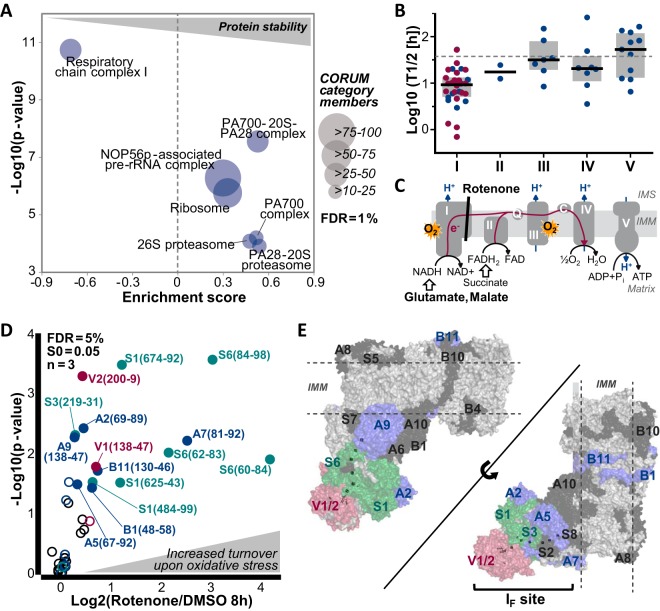
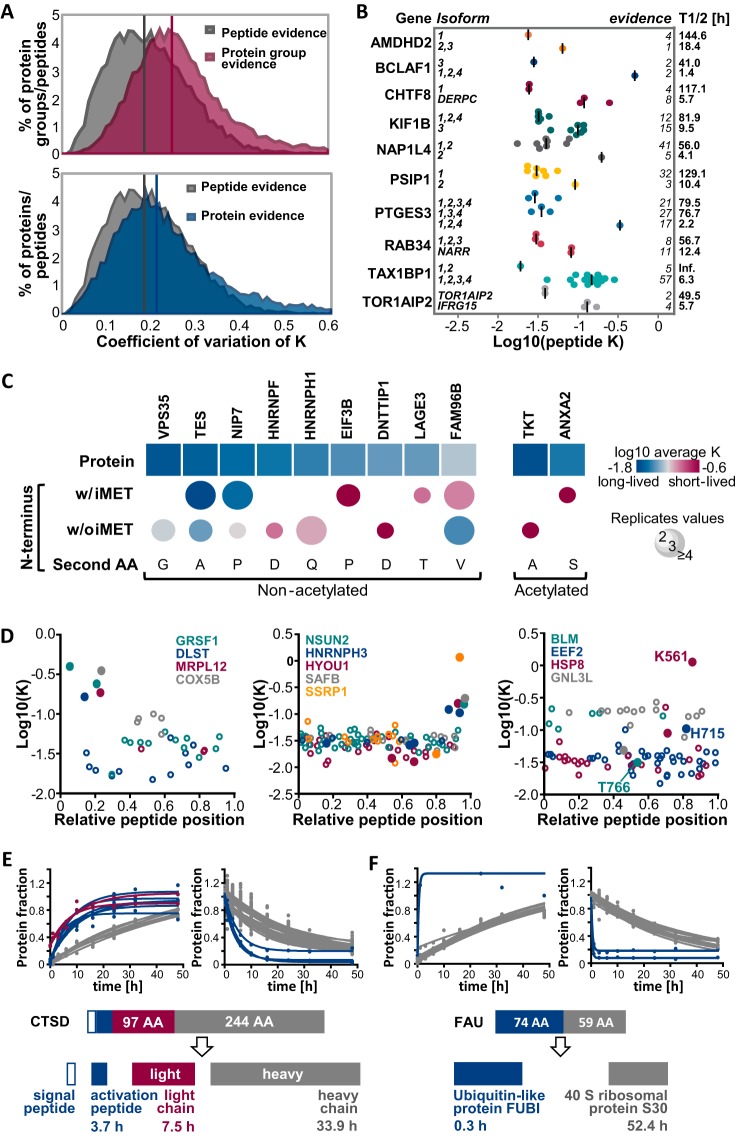
The coordination of protein synthesis and degradation regulating protein abundance is a fundamental process in cellular homeostasis. Today, mass spectrometry-based technologies allow determination of endogenous protein turnover on a proteome-wide scale. However, standard dynamic SILAC (Stable Isotope Labeling in Cell Culture) approaches can suffer from missing data across pulse time-points limiting the accuracy of such analysis. This issue is of particular relevance when studying protein stability at the level of proteoforms because often only single peptides distinguish between different protein products of the same gene. To address this shortcoming, we evaluated the merits of combining dynamic SILAC and tandem mass tag (TMT)-labeling of ten pulse time-points in a single experiment. Although the comparison to the standard dynamic SILAC method showed a high concordance of protein turnover rates, the pulsed SILAC-TMT approach yielded more comprehensive data (6000 proteins on average) without missing values. Replicate analysis further established that the same reproducibility of turnover rate determination can be obtained for peptides and proteins facilitating proteoform resolved investigation of protein stability. We provide several examples of differentially turned over splice variants and show that post-translational modifications can affect cellular protein half-lives. For example, N-terminally processed peptides exhibited both faster and slower turnover behavior compared with other peptides of the same protein. In addition, the suspected proteolytic processing of the fusion protein FAU was substantiated by measuring vastly different stabilities of the cleavage products. Furthermore, differential peptide turnover suggested a previously unknown mechanism of activity regulation by post-translational destabilization of cathepsin D as well as the DNA helicase BLM. Finally, our comprehensive data set facilitated a detailed evaluation of the impact of protein properties and functions on protein stability in steady-state cells and uncovered that the high turnover of respiratory chain complex I proteins might be explained by oxidative stress.
© 2018 Zecha et al.
Figures
References
-
- Wilhelm M., Schlegl J., Hahne H., Gholami A. M., Lieberenz M., Savitski M. M., Ziegler E., Butzmann L., Gessulat S., Marx H., Mathieson T., Lemeer S., Schnatbaum K., Reimer U., Wenschuh H., Mollenhauer M., Slotta-Huspenina J., Boese J.-H., Bantscheff M., Gerstmair A., Faerber F., and Kuster B. (2014) Mass-spectrometry-based draft of the human proteome. Nature 509, 582. - PubMed
-
- White A. (1942) The Dynamic State of Body Constituents. Yale J. Biol. Med. 14, 677–677
-
- Hinkson I. V., and Elias J. E. (2011) The dynamic state of protein turnover: It's about time. Trends Cell Biol. 21, 293–303 - PubMed
-
- Wheatley D. N., Giddings M. R., and Inglis M. S. (1980) Kinetics of degradation of ‘short-’ and ‘long-lived’ proteins in cultured mammalian cells. Cell Biol. Int. Reports 4, 1081–1090 - PubMed
-
- Dittrich A., Siewert E., and Schaper F. (2013) Determination of Protein Turnover Rates in the JAK/STAT Pathway Using a Radioactive Pulse-Chase Approach. In: Nicholson S. E., and Nicola N. A., eds. JAK-STAT Signalling: Methods and Protocols, pp. 69–80, Humana Press, Totowa, NJ - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
