

A computational approach to distinguish somatic vs. germline origin of genomic alterations from deep sequencing of cancer specimens without a matched normal
- PMID: 29415044
- PMCID: PMC5832436
- DOI: 10.1371/journal.pcbi.1005965
A computational approach to distinguish somatic vs. germline origin of genomic alterations from deep sequencing of cancer specimens without a matched normal
Abstract
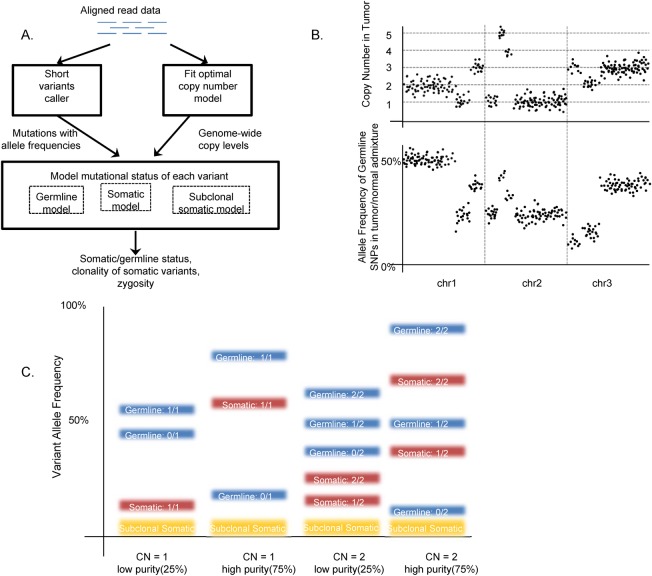
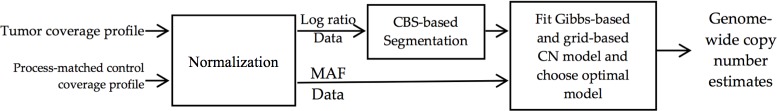
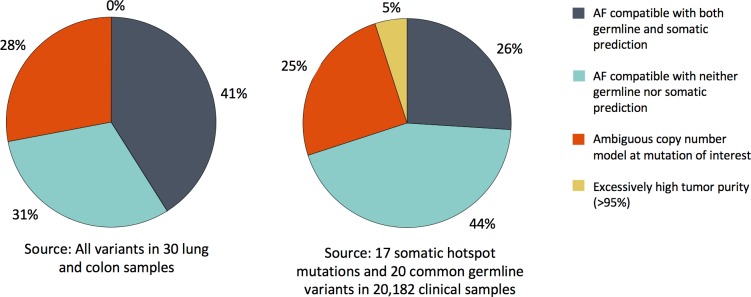
A key constraint in genomic testing in oncology is that matched normal specimens are not commonly obtained in clinical practice. Thus, while well-characterized genomic alterations do not require normal tissue for interpretation, a significant number of alterations will be unknown in whether they are germline or somatic, in the absence of a matched normal control. We introduce SGZ (somatic-germline-zygosity), a computational method for predicting somatic vs. germline origin and homozygous vs. heterozygous or sub-clonal state of variants identified from deep massively parallel sequencing (MPS) of cancer specimens. The method does not require a patient matched normal control, enabling broad application in clinical research. SGZ predicts the somatic vs. germline status of each alteration identified by modeling the alteration's allele frequency (AF), taking into account the tumor content, tumor ploidy, and the local copy number. Accuracy of the prediction depends on the depth of sequencing and copy number model fit, which are achieved in our clinical assay by sequencing to high depth (>500x) using MPS, covering 394 cancer-related genes and over 3,500 genome-wide single nucleotide polymorphisms (SNPs). Calls are made using a statistic based on read depth and local variability of SNP AF. To validate the method, we first evaluated performance on samples from 30 lung and colon cancer patients, where we sequenced tumors and matched normal tissue. We examined predictions for 17 somatic hotspot mutations and 20 common germline SNPs in 20,182 clinical cancer specimens. To assess the impact of stromal admixture, we examined three cell lines, which were titrated with their matched normal to six levels (10-75%). Overall, predictions were made in 85% of cases, with 95-99% of variants predicted correctly, a significantly superior performance compared to a basic approach based on AF alone. We then applied the SGZ method to the COSMIC database of known somatic variants in cancer and found >50 that are in fact more likely to be germline.
Conflict of interest statement
I have read the journal's policy and the authors of this manuscript have the following competing interests: JXS, YH, MM, GMF, JSR, VAM, PJS, and DL are paid employees of Foundation Medicine. JXS, YH, ES, GMF, JSR, VAM, PJS, DL, and RY are shareholders of Foundation Medicine. ES and RY are former employees of Foundation Medicine, but were employees when the studies in this paper were conducted.
Figures
References
-
- Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31(11):1023–31. doi: 10.1038/nbt.2696 - DOI - PMC - PubMed
-
- Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486(7403):400–4. doi: 10.1038/nature11017 - DOI - PMC - PubMed
-
- Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513(7517):202–9. doi: 10.1038/nature13480 - DOI - PMC - PubMed
-
- Kanchi KL, Johnson KJ, Lu C, McLellan MD, Leiserson MD, Wendl MC, et al. Integrated analysis of germline and somatic variants in ovarian cancer. Nat Commun. 2014;5:3156 doi: 10.1038/ncomms4156 - DOI - PMC - PubMed
-
- Koboldt DC, Steinberg KM, Larson DE, Wilson RK, Mardis ER. The next-generation sequencing revolution and its impact on genomics. Cell. 2013;155(1):27–38. doi: 10.1016/j.cell.2013.09.006 - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
