

Chronic cigarette smoke exposure induces systemic hypoxia that drives intestinal dysfunction
- PMID: 29415878
- PMCID: PMC5821186
- DOI: 10.1172/jci.insight.94040
Chronic cigarette smoke exposure induces systemic hypoxia that drives intestinal dysfunction
Abstract
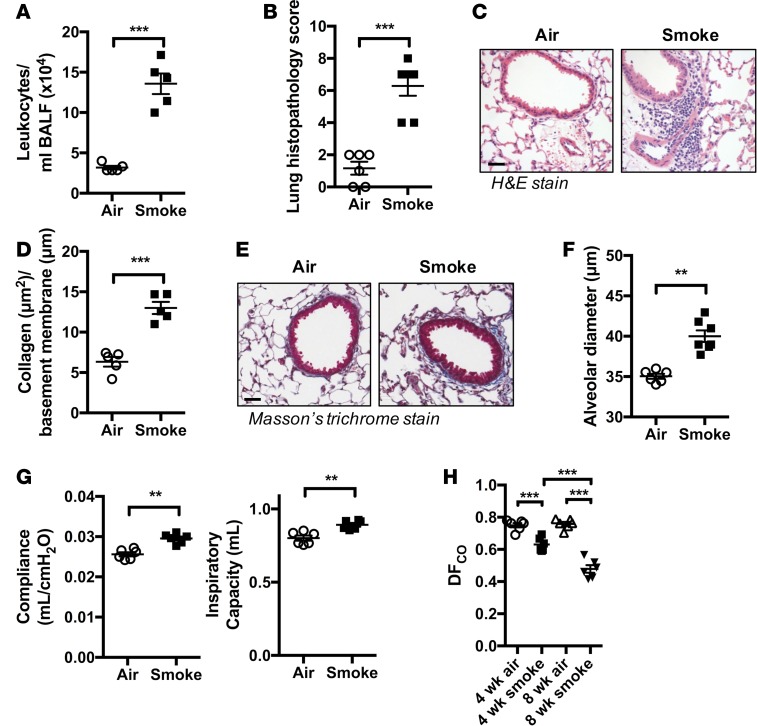
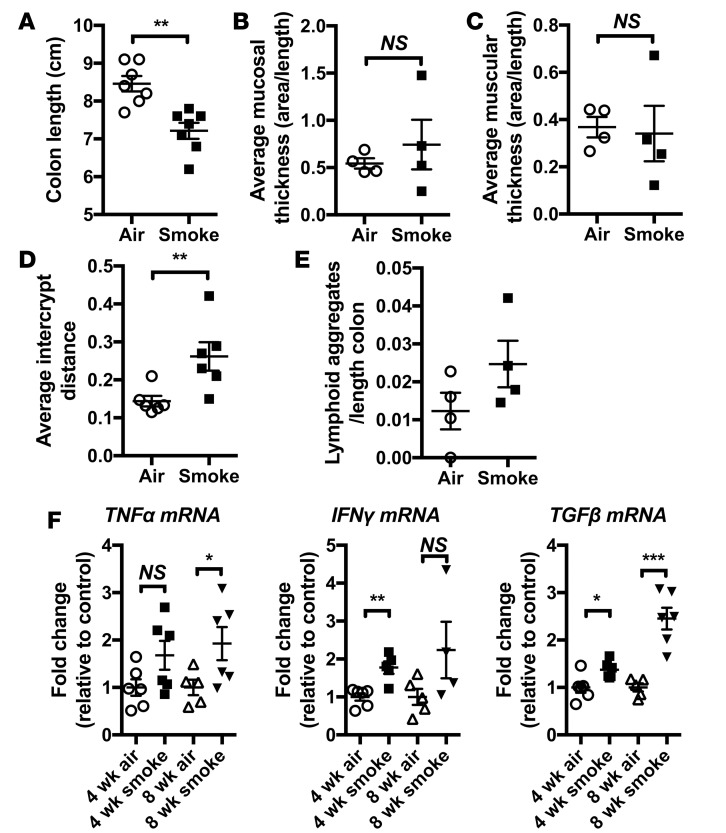
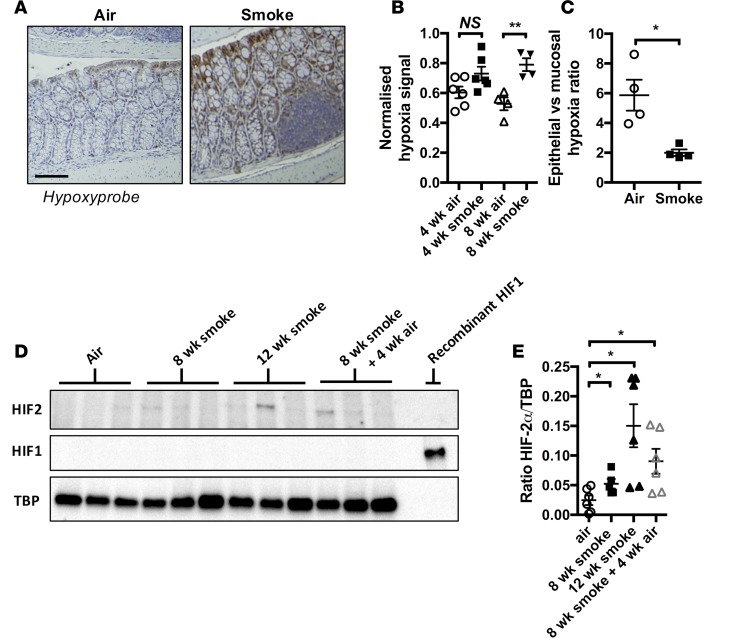
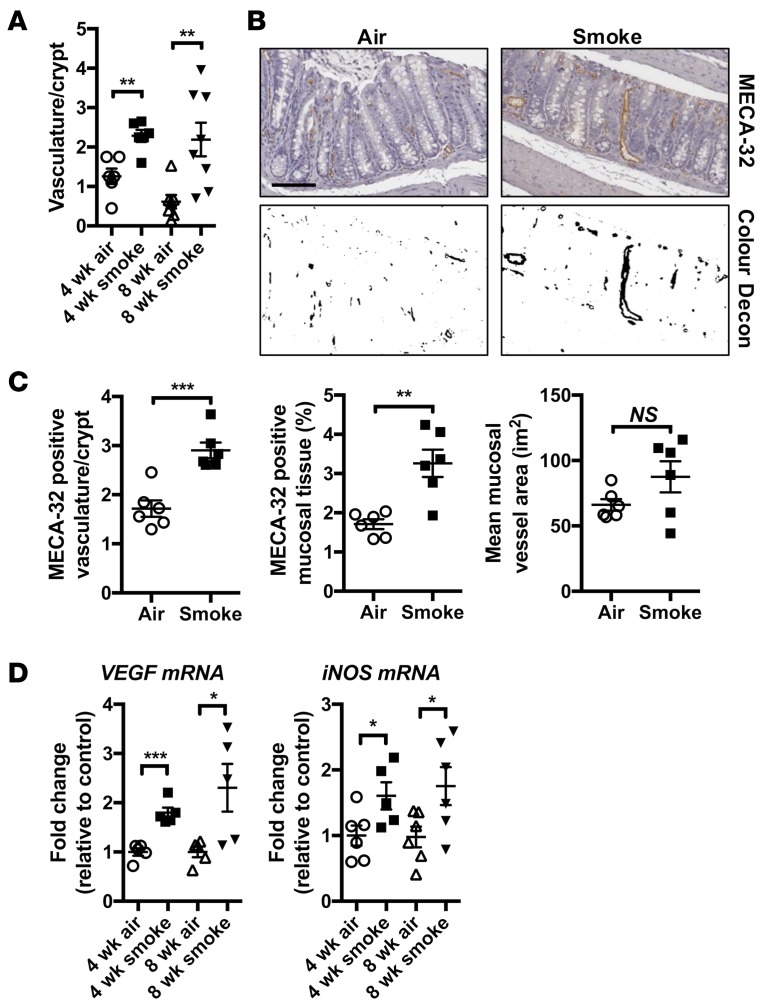
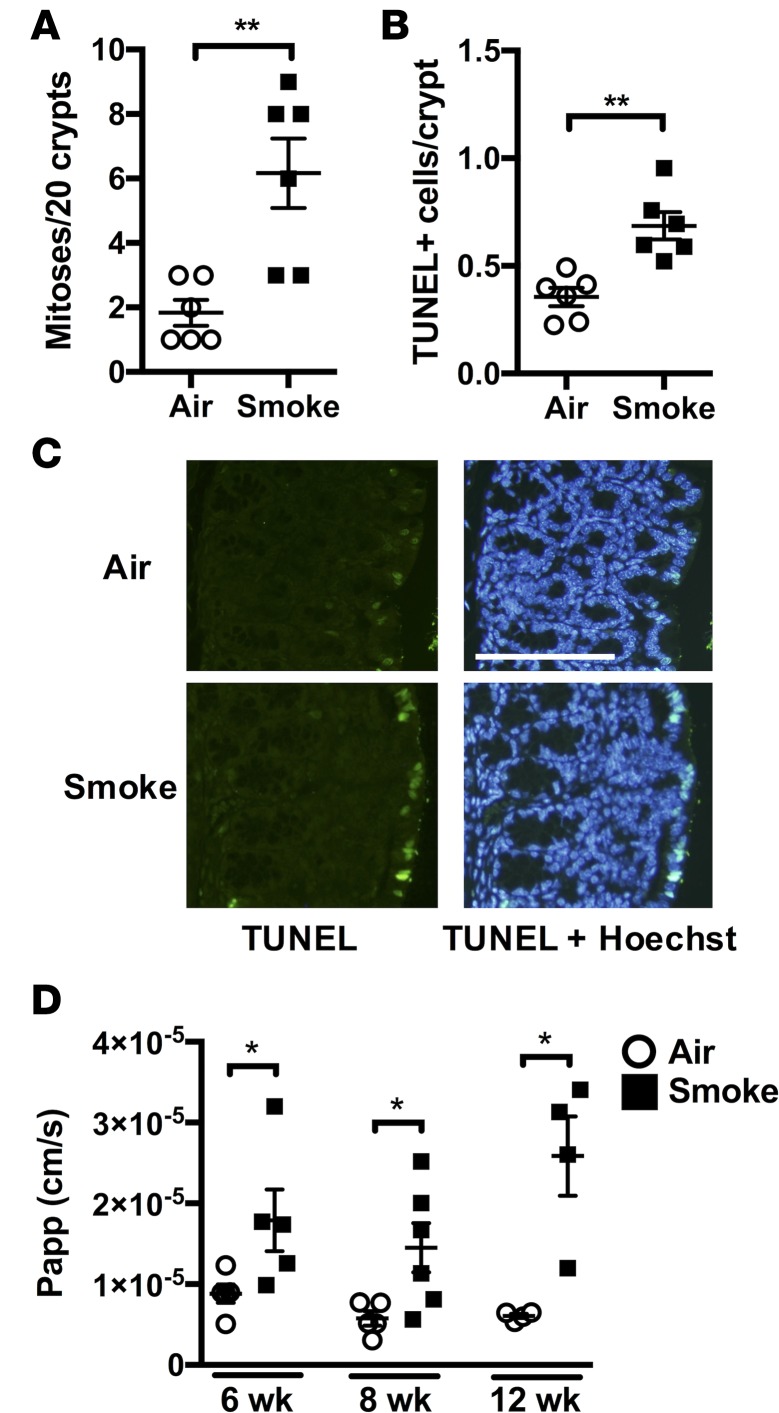
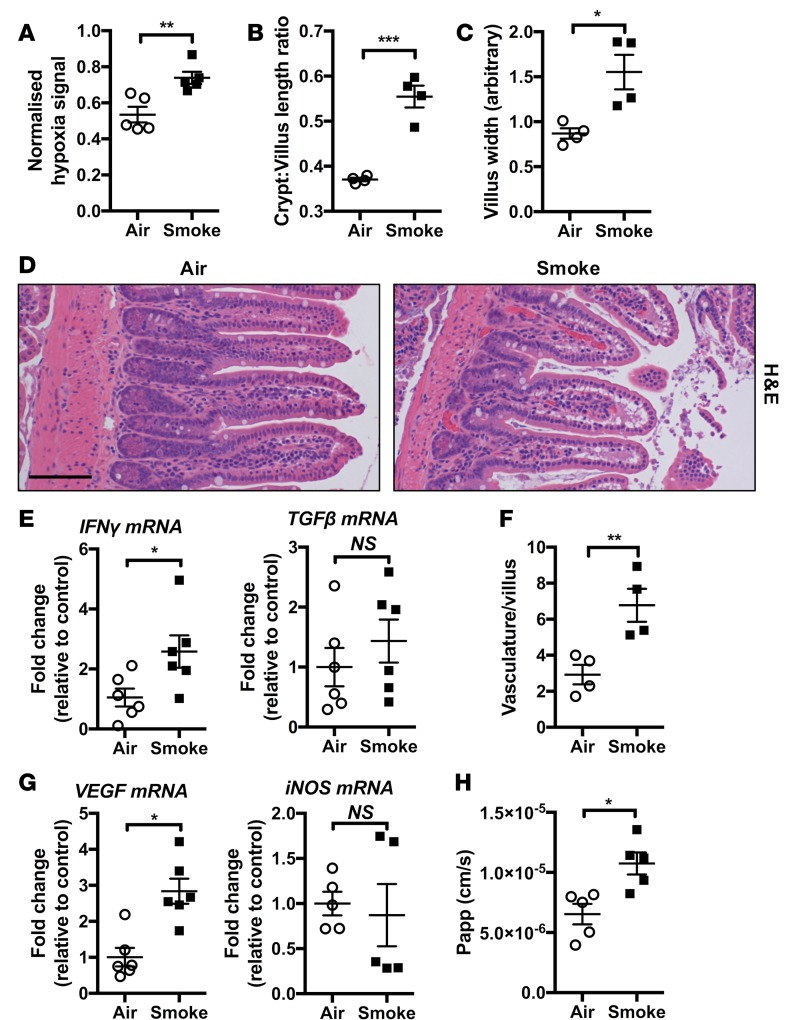
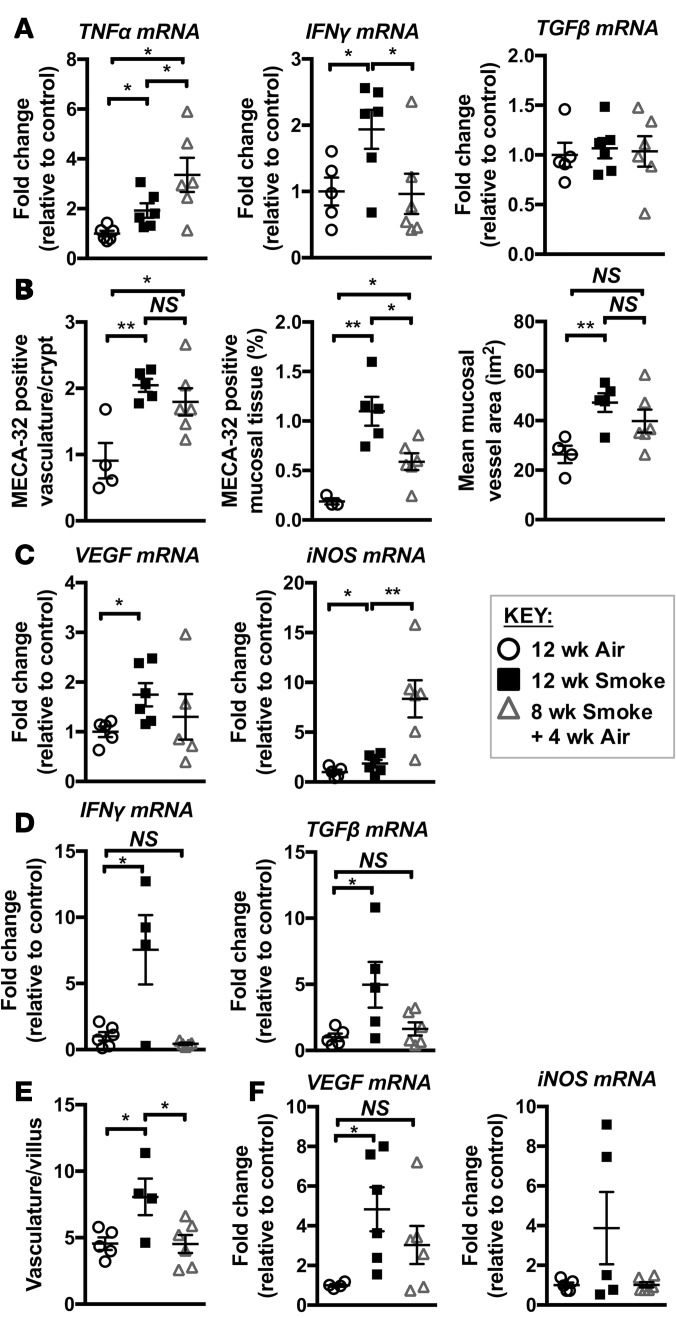
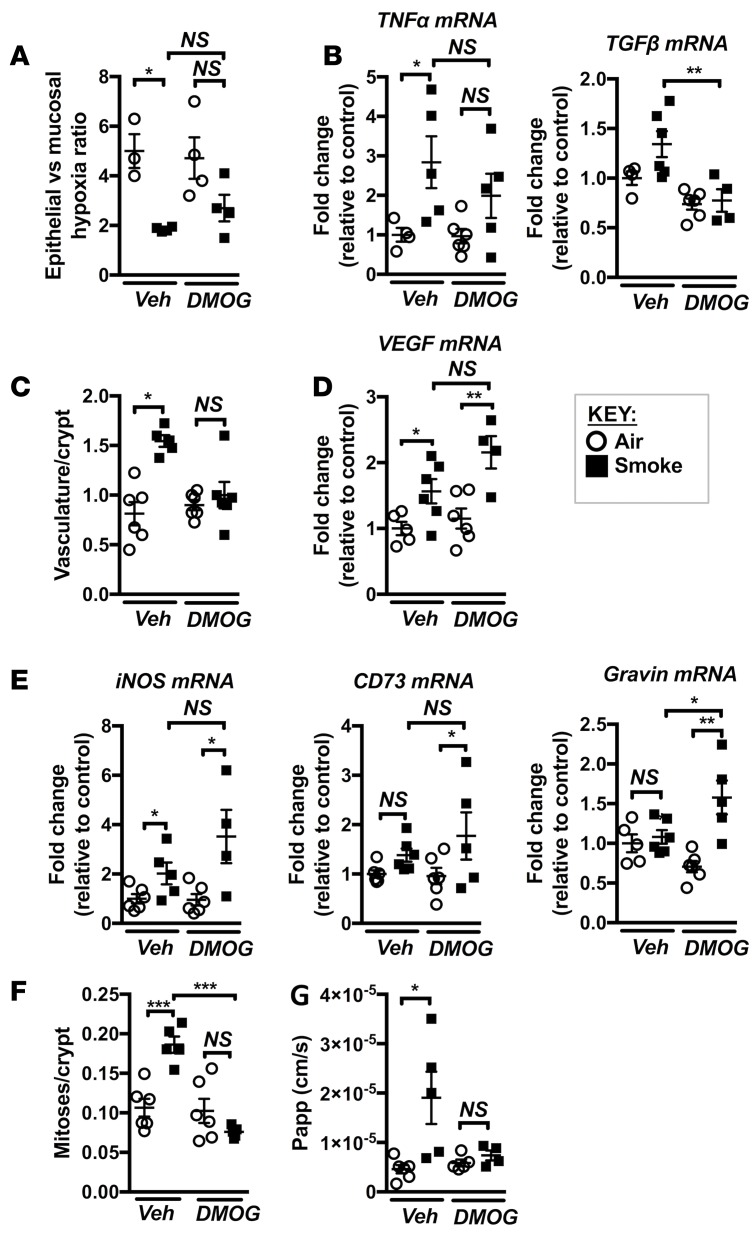
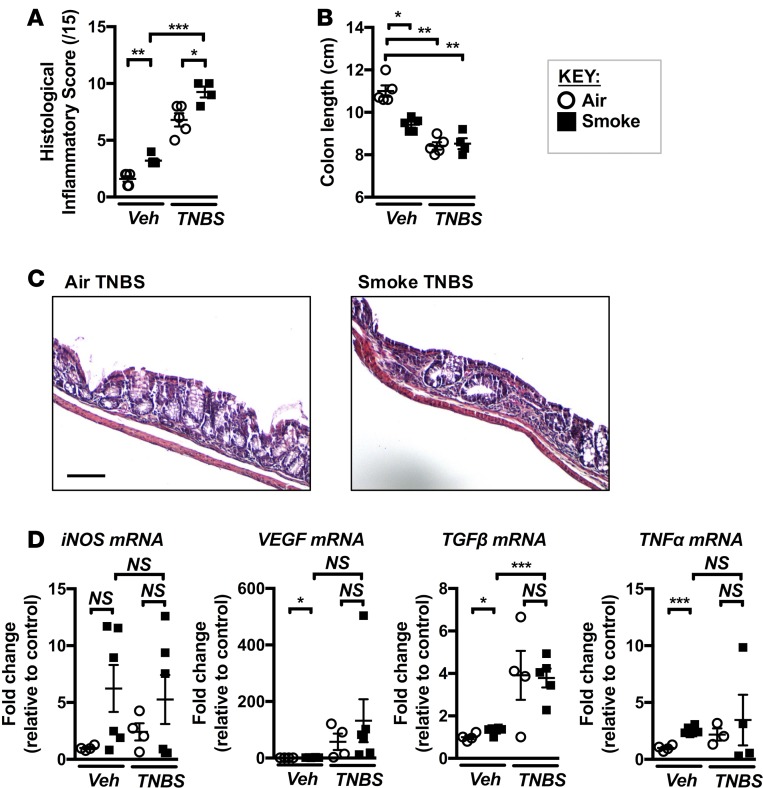
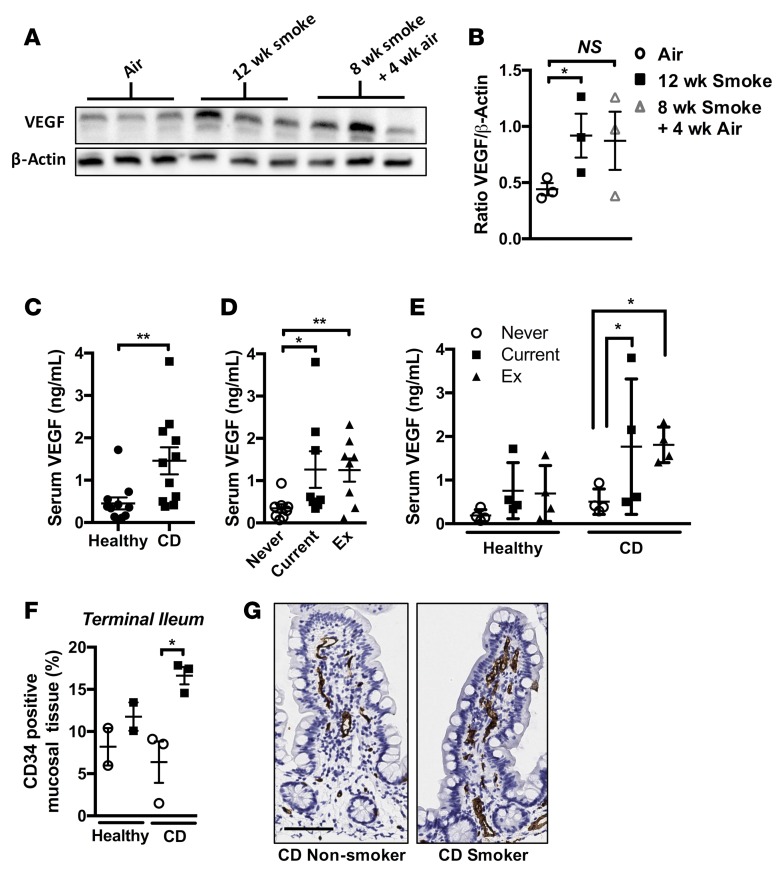
Crohn's disease (CD) is a chronic inflammatory disease of the gastrointestinal tract (GIT). Cigarette smoke (CS) exposure and chronic obstructive pulmonary disease (COPD) are risk factors for CD, although the mechanisms involved are poorly understood. We employed a mouse model of CS-induced experimental COPD and clinical studies to examine these mechanisms. Concurrent with the development of pulmonary pathology and impaired gas exchange, CS-exposed mice developed CD-associated pathology in the colon and ileum, including gut mucosal tissue hypoxia, HIF-2 stabilization, inflammation, increased microvasculature, epithelial cell turnover, and decreased intestinal barrier function. Subsequent smoking cessation reduced GIT pathology, particularly in the ileum. Dimethyloxaloylglycine, a pan-prolyl hydroxylase inhibitor, ameliorated CS-induced GIT pathology independently of pulmonary pathology. Prior smoke exposure exacerbated intestinal pathology in 2,4,6-trinitrobenzenesulfonic acid-induced (TNBS-induced) colitis. Circulating vascular endothelial growth factor, a marker of systemic hypoxia, correlated with CS exposure and CD in mice and humans. Increased mucosal vascularisation was evident in ileum biopsies from CD patients who smoke compared with nonsmokers, supporting our preclinical data. We provide strong evidence that chronic CS exposure and, for the first time to our knowledge, associated impaired gas exchange cause systemic and intestinal ischemia, driving angiogenesis and GIT epithelial barrier dysfunction, resulting in increased risk and severity of CD.
Keywords: COPD; Gastroenterology; Inflammatory bowel disease; Pulmonology; hypoxia.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
