

Lysogeny is prevalent and widely distributed in the murine gut microbiota
- PMID: 29416123
- PMCID: PMC5864201
- DOI: 10.1038/s41396-018-0061-9
Lysogeny is prevalent and widely distributed in the murine gut microbiota
Abstract
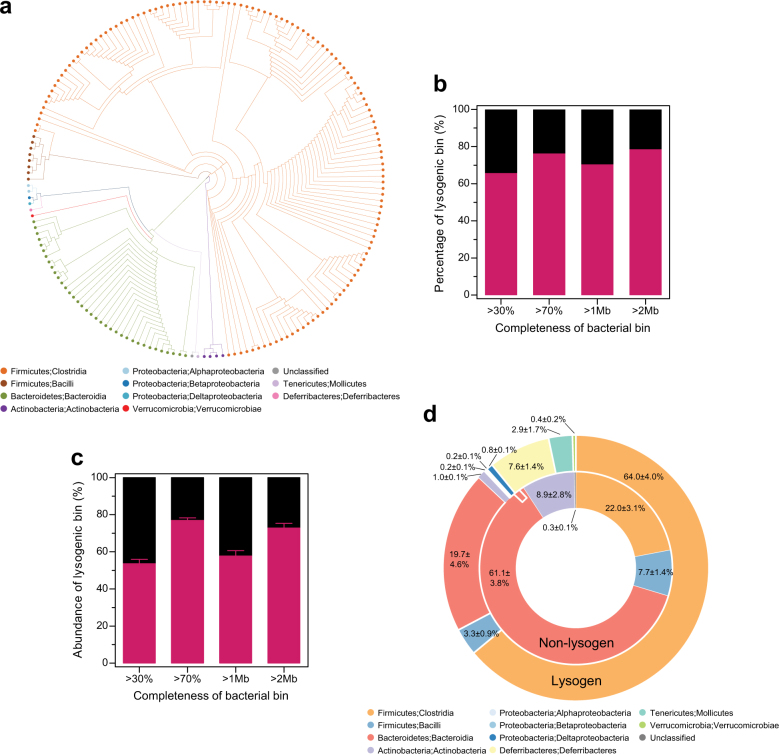
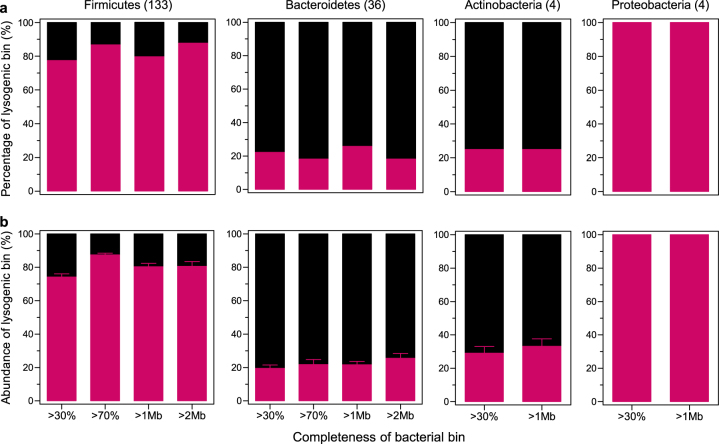
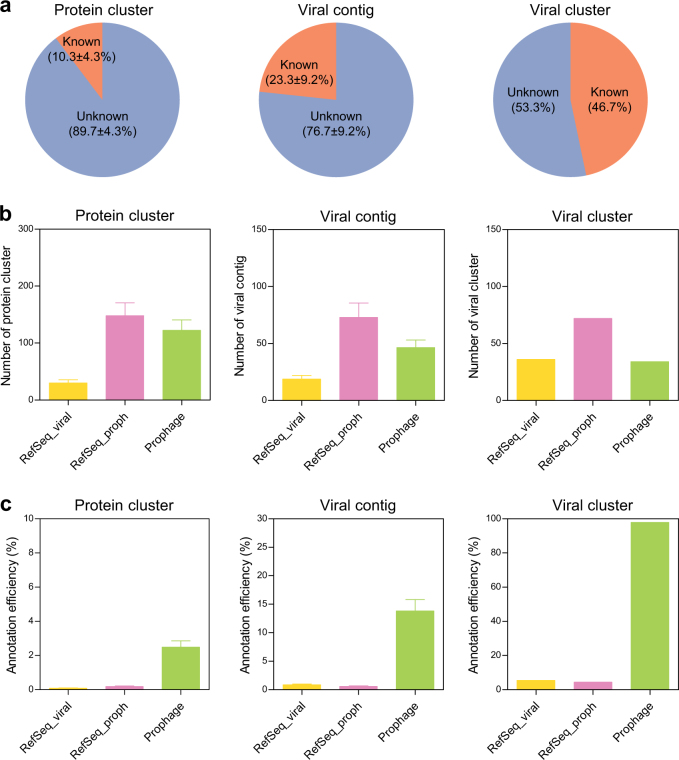
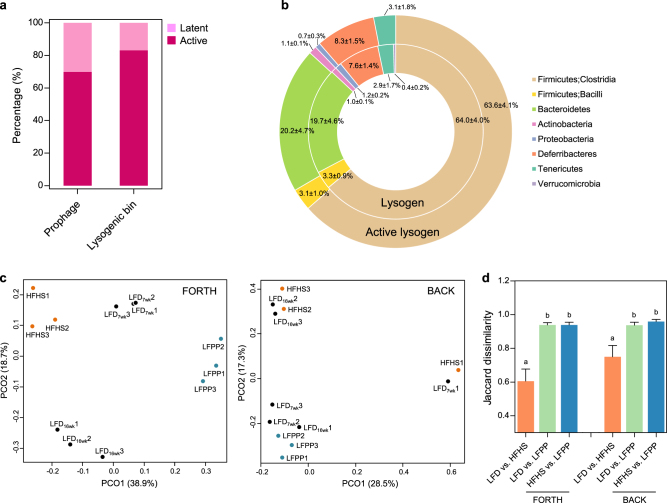
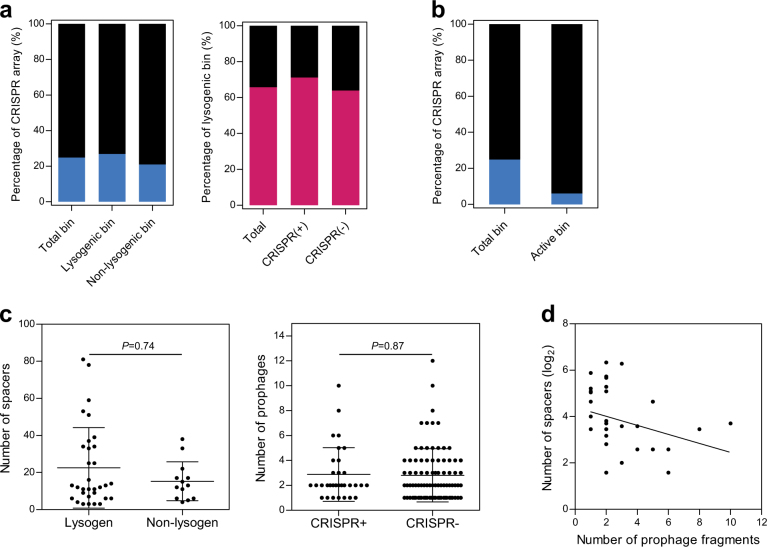
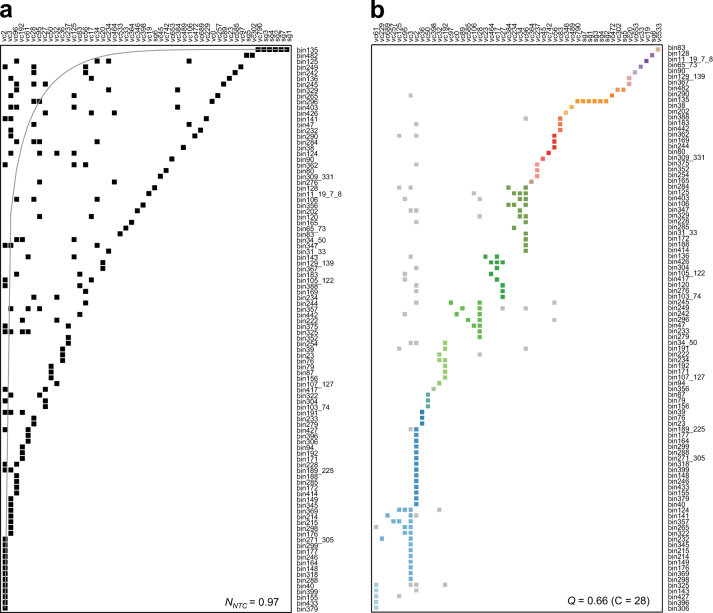
Bacteriophages are central members and potential modulators of the gut microbiome; however, the ecological and evolutionary relationships of gut bacteria and phages are poorly understood. Here we investigated the abundance and diversity of lysogenic bacteria (lysogens) in the bacterial community of C57BL/6J mice by detecting integrated prophages in genomes reconstructed from the metagenome of commensal bacteria. For the activities of lysogens and prophages, we compared the prophage genomes with the metagenome of free phages. The majority of commensal bacteria in different taxa were identified as lysogens. More lysogens were found among Firmicutes and Proteobacteria, than among Bacteroidetes and Actinobacteria. The prophage genomes shared high sequence similarity with the metagenome of free phages, indicating that most lysogens appeared to be active, and that prophages are spontaneously induced as active phages; dietary interventions changed the composition of the induced prophages. By contrast, CRISPR-Cas systems were present in few commensal bacteria, and were rarely active against gut phages. The structure of the bacteria-phage infection networks was "nested-modular", with modularity emerging across taxonomic scales, indicating that temperate phage features have developed over a long phylogenetic timescale. We concluded that phage generalists contribute to the prevalence of lysogeny in the gut ecosystem.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures

References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
