

Regulation of mutant TERT by BRAF V600E/MAP kinase pathway through FOS/GABP in human cancer
- PMID: 29422527
- PMCID: PMC5805723
- DOI: 10.1038/s41467-018-03033-1
Regulation of mutant TERT by BRAF V600E/MAP kinase pathway through FOS/GABP in human cancer
Abstract
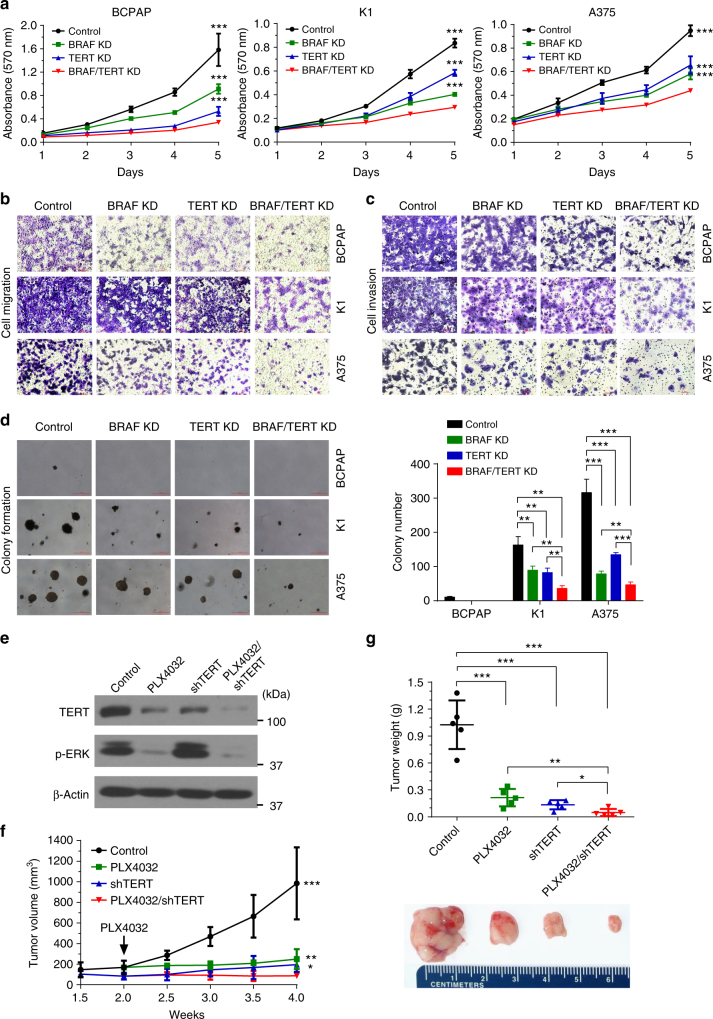
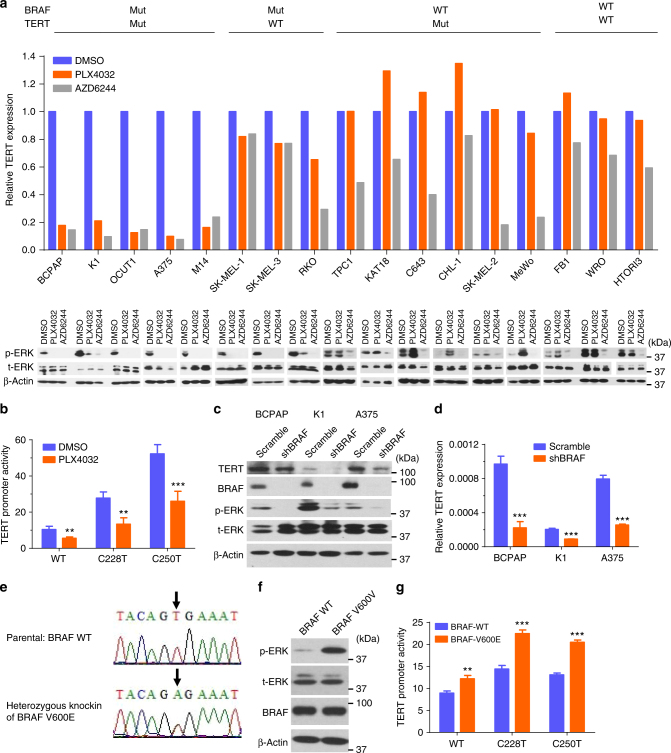
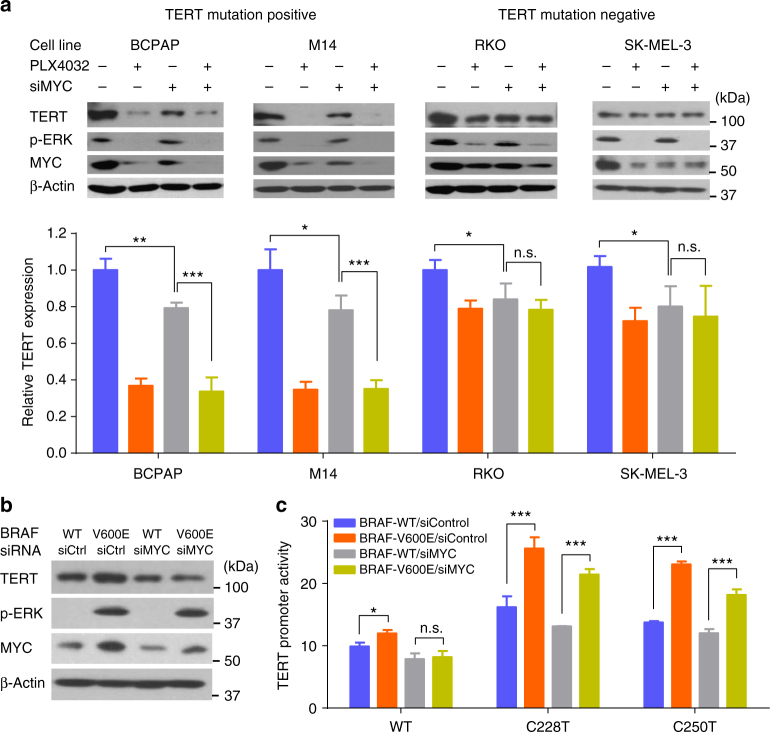
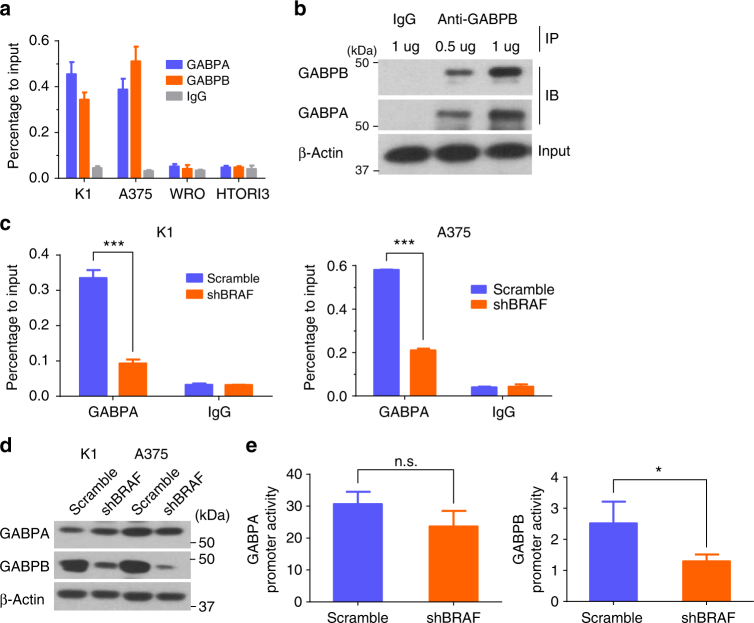
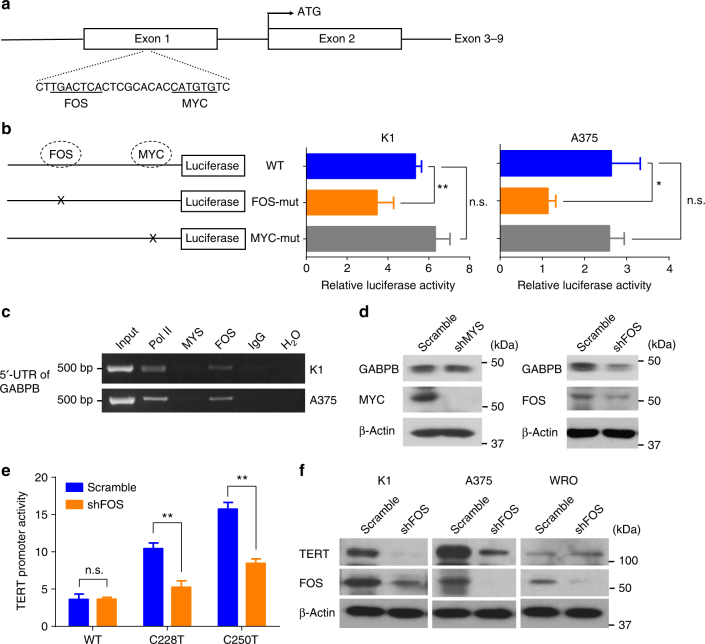
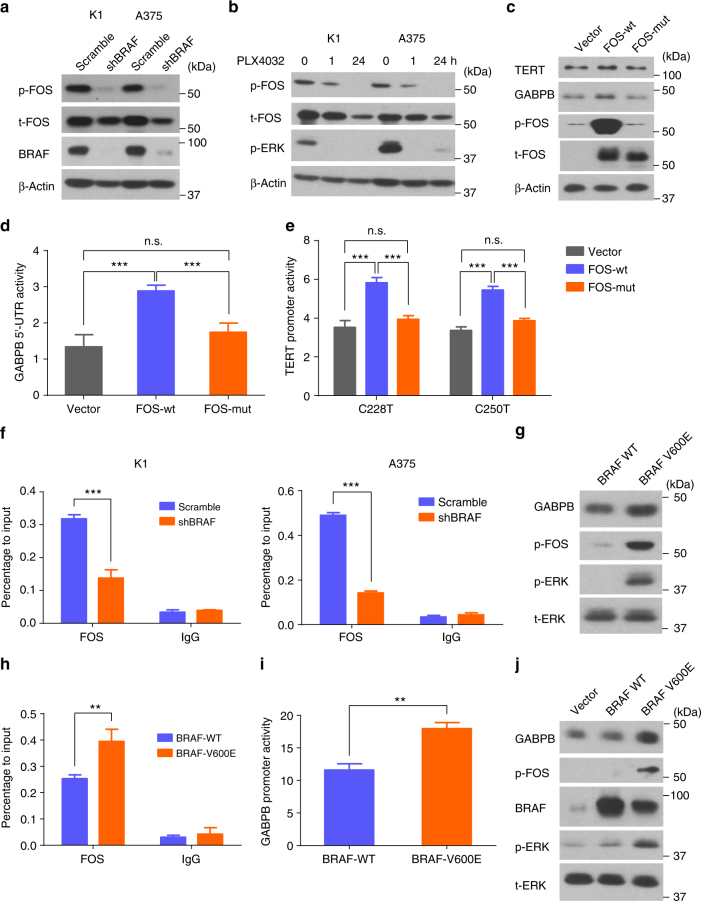
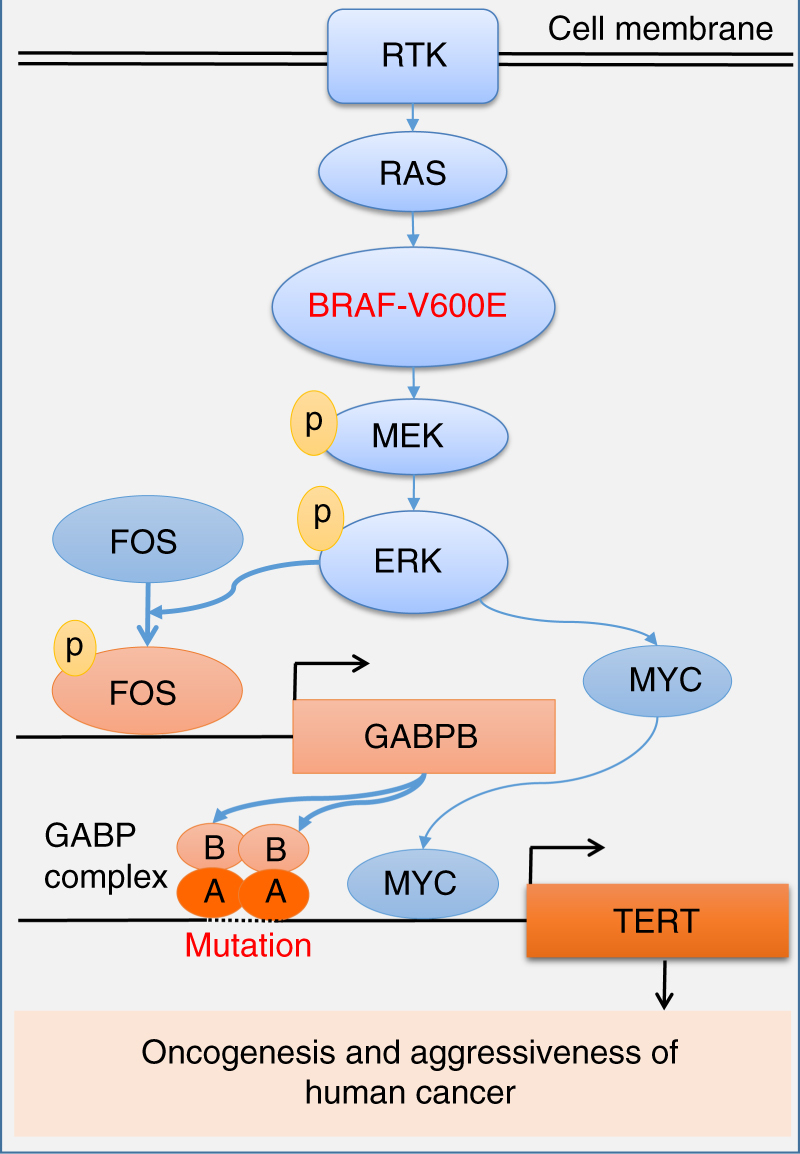
The unique oncogene duet of coexisting BRAF V600E and TERT promoter mutations are widely found to be a robust genetic background promoting human cancer aggressiveness, but the mechanism is unclear. Here, we demonstrate that the BRAF V600E/MAP kinase pathway phosphorylates and activates FOS, which in turn acts as a transcription factor to bind and activate the GABPB promoter, increasing GABPB expression and driving formation of GABPA-GABPB complex; the latter selectively binds and activates mutant TERT promoter, upregulating TERT expression. Elevated TERT functions as a strong oncoprotein, robustly promoting aggressive behaviors of cancer cells and tumor development. We thus identify a molecular mechanism for the activation of mutant TERT by the BRAF V600E/MAP kinase pathway, in which FOS as a transcriptional factor of GABPB promoter plays a key role in functionally bridging the two oncogenes in cooperatively promoting oncogenesis, providing important cancer biological and clinical implications.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
