

Mutations in the BAF-Complex Subunit DPF2 Are Associated with Coffin-Siris Syndrome
- PMID: 29429572
- PMCID: PMC5985265
- DOI: 10.1016/j.ajhg.2018.01.014
Mutations in the BAF-Complex Subunit DPF2 Are Associated with Coffin-Siris Syndrome
Abstract
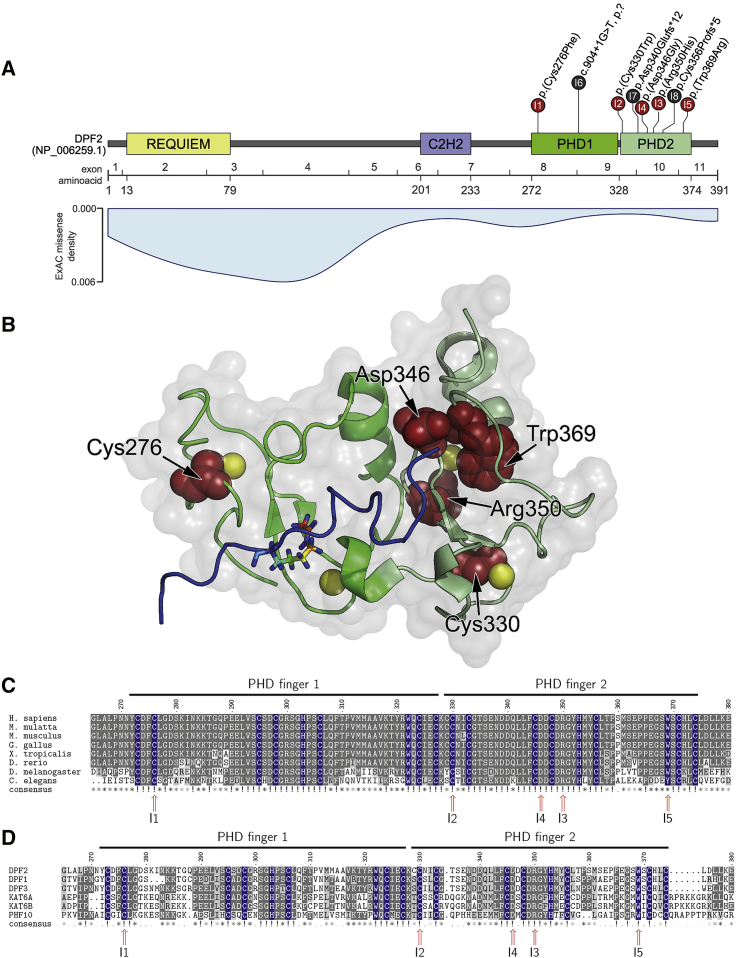
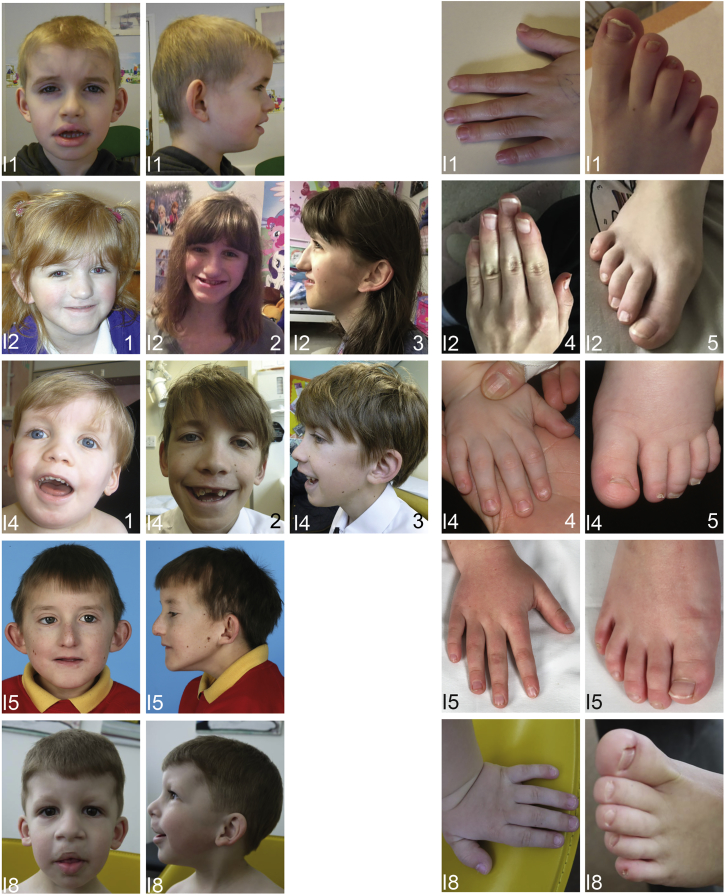
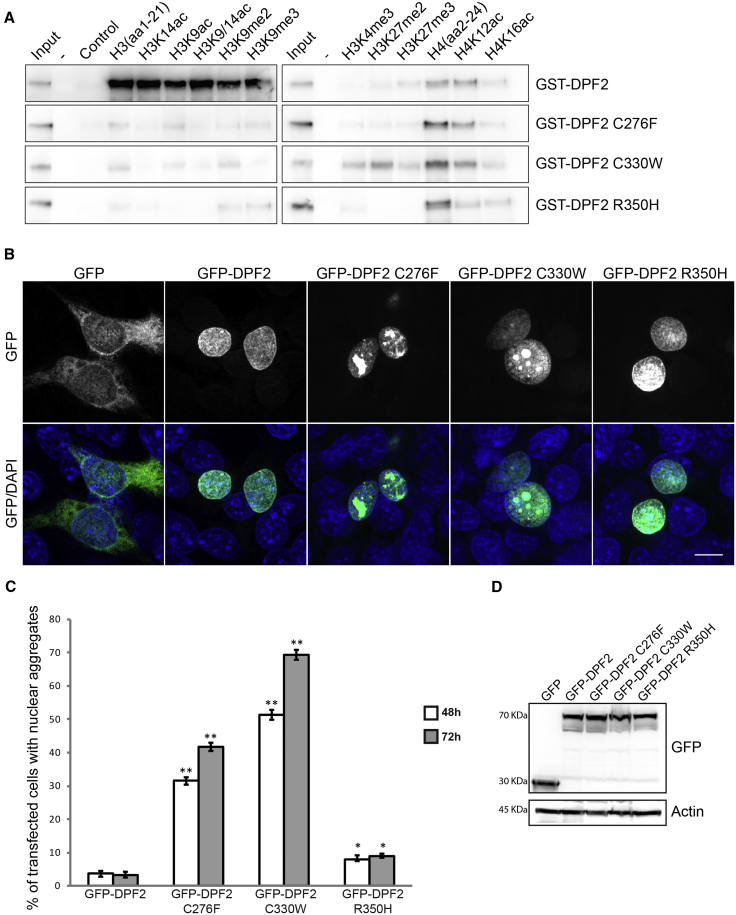
Variants affecting the function of different subunits of the BAF chromatin-remodelling complex lead to various neurodevelopmental syndromes, including Coffin-Siris syndrome. Furthermore, variants in proteins containing PHD fingers, motifs recognizing specific histone tail modifications, have been associated with several neurological and developmental-delay disorders. Here, we report eight heterozygous de novo variants (one frameshift, two splice site, and five missense) in the gene encoding the BAF complex subunit double plant homeodomain finger 2 (DPF2). Affected individuals share common clinical features described in individuals with Coffin-Siris syndrome, including coarse facial features, global developmental delay, intellectual disability, speech impairment, and hypoplasia of fingernails and toenails. All variants occur within the highly conserved PHD1 and PHD2 motifs. Moreover, missense variants are situated close to zinc binding sites and are predicted to disrupt these sites. Pull-down assays of recombinant proteins and histone peptides revealed that a subset of the identified missense variants abolish or impaire DPF2 binding to unmodified and modified H3 histone tails. These results suggest an impairment of PHD finger structural integrity and cohesion and most likely an aberrant recognition of histone modifications. Furthermore, the overexpression of these variants in HEK293 and COS7 cell lines was associated with the formation of nuclear aggregates and the recruitment of both wild-type DPF2 and BRG1 to these aggregates. Expression analysis of truncating variants found in the affected individuals indicated that the aberrant transcripts escape nonsense-mediated decay. Altogether, we provide compelling evidence that de novo variants in DPF2 cause Coffin-Siris syndrome and propose a dominant-negative mechanism of pathogenicity.
Keywords: BAF complex; Coffin-Siris syndrome; DPF2; PHD finger; autism spectrum disorder; dominant negative; histone modification; intellectual disability; nail hypoplasia; nuclear aggregates.
Copyright © 2018 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Kosho T., Okamoto N., Ohashi H., Tsurusaki Y., Imai Y., Hibi-Ko Y., Kawame H., Homma T., Tanabe S., Kato M. Clinical correlations of mutations affecting six components of the SWI/SNF complex: detailed description of 21 patients and a review of the literature. Am. J. Med. Genet. A. 2013;161A:1221–1237. - PubMed
-
- Coffin G.S., Siris E. Mental retardation with absent fifth fingernail and terminal phalanx. Am. J. Dis. Child. 1970;119:433–439. - PubMed
-
- Kosho T., Miyake N., Carey J.C. Coffin-Siris syndrome and related disorders involving components of the BAF (mSWI/SNF) complex: historical review and recent advances using next generation sequencing. Am. J. Med. Genet. C. Semin. Med. Genet. 2014;166C:241–251. - PubMed
-
- Kosho T., Okamoto N., Coffin-Siris Syndrome International Collaborators Genotype-phenotype correlation of Coffin-Siris syndrome caused by mutations in SMARCB1, SMARCA4, SMARCE1, and ARID1A. Am. J. Med. Genet. C. Semin. Med. Genet. 2014;166C:262–275. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
