

The Personal Genome Project Canada: findings from whole genome sequences of the inaugural 56 participants
- PMID: 29431110
- PMCID: PMC5798982
- DOI: 10.1503/cmaj.171151
The Personal Genome Project Canada: findings from whole genome sequences of the inaugural 56 participants
Abstract
Background: The Personal Genome Project Canada is a comprehensive public data resource that integrates whole genome sequencing data and health information. We describe genomic variation identified in the initial recruitment cohort of 56 volunteers.
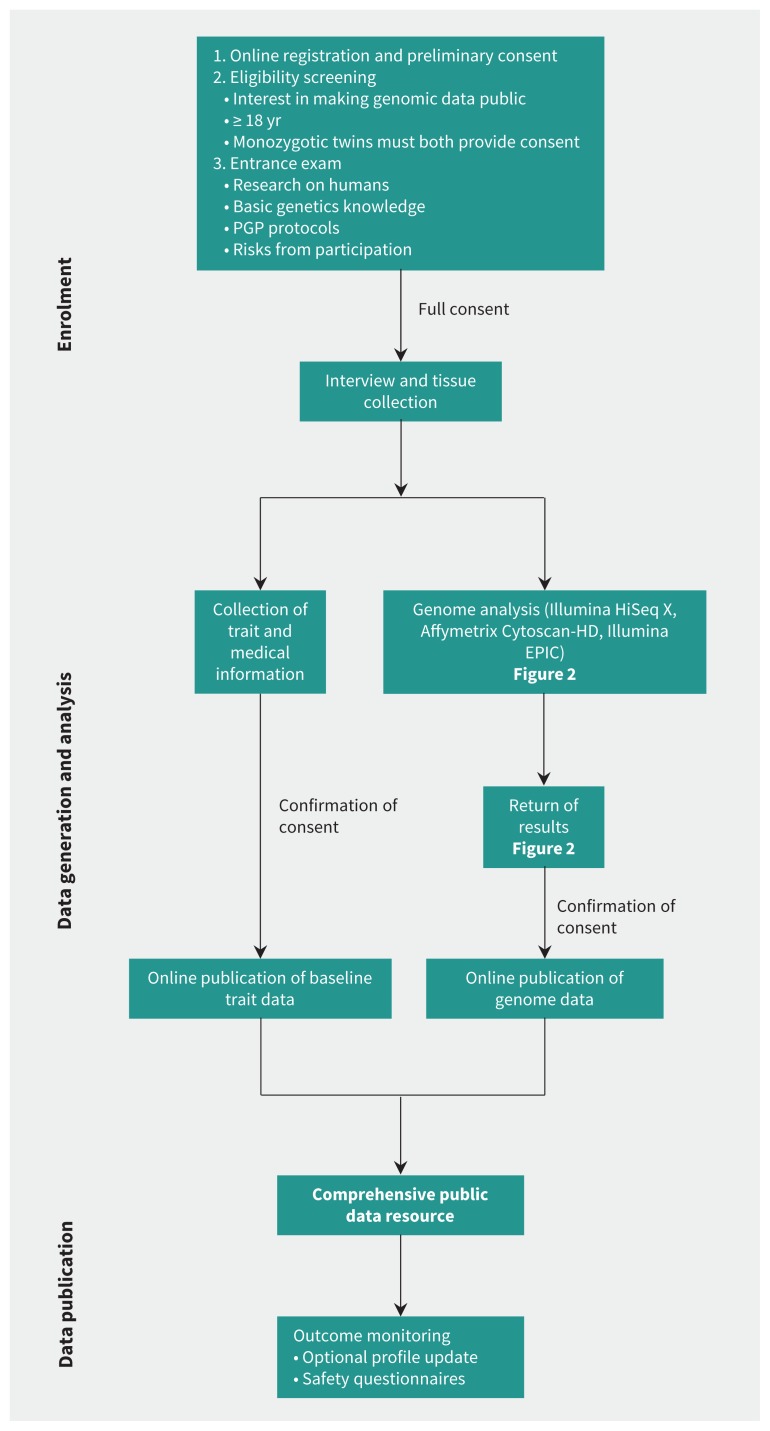
Methods: Volunteers were screened for eligibility and provided informed consent for open data sharing. Using blood DNA, we performed whole genome sequencing and identified all possible classes of DNA variants. A genetic counsellor explained the implication of the results to each participant.
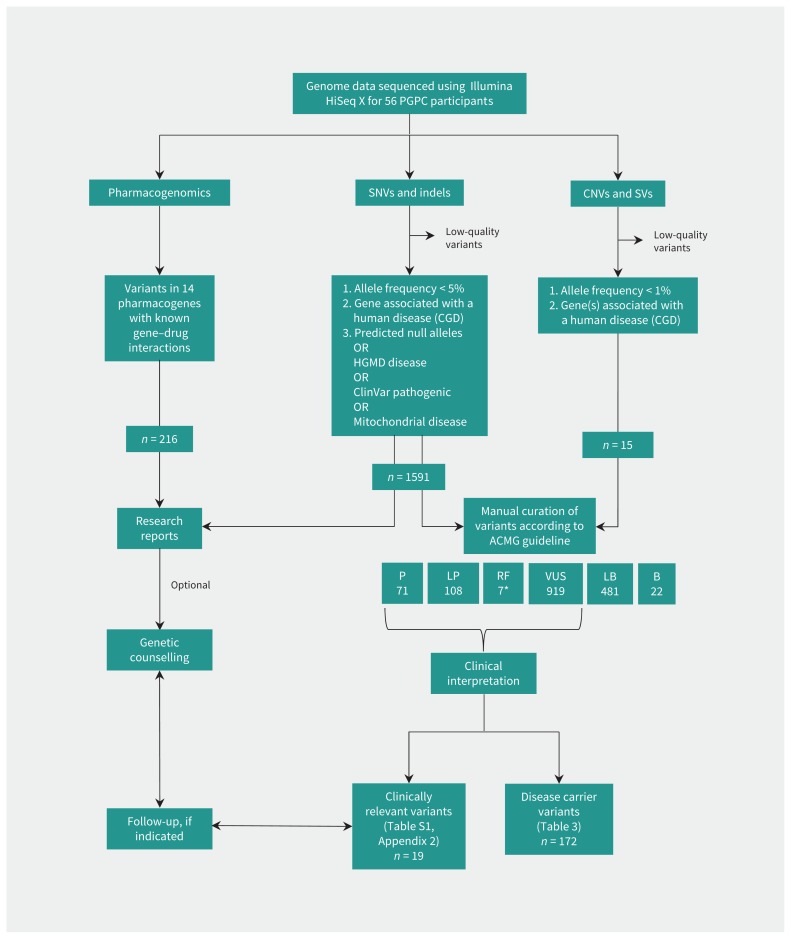
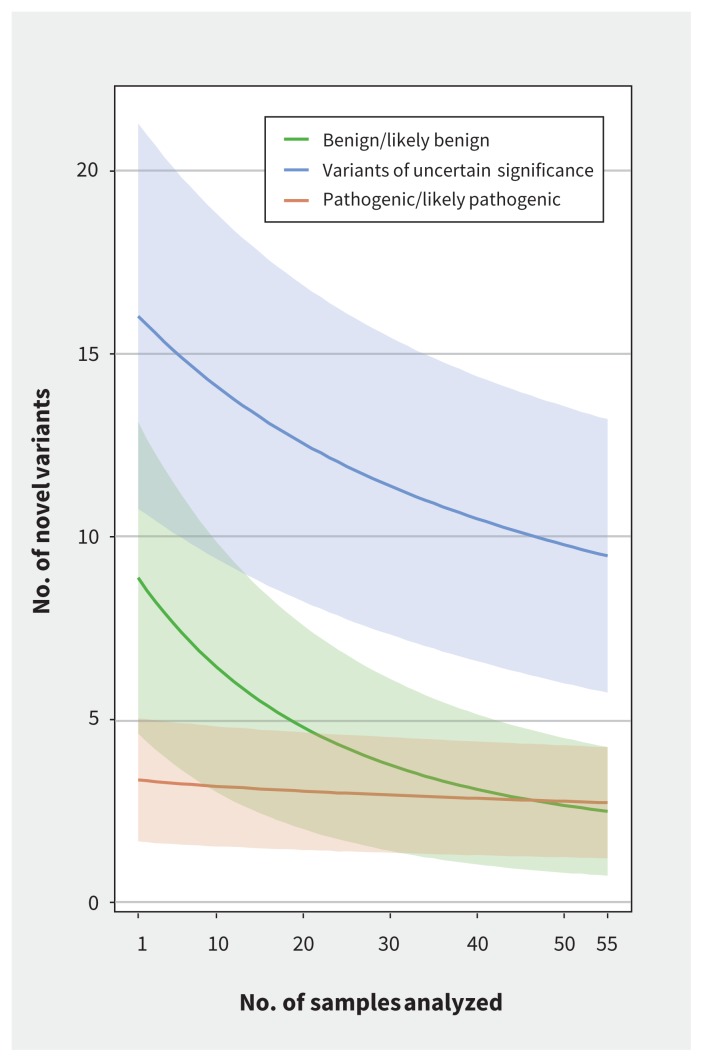
Results: Whole genome sequencing of the first 56 participants identified 207 662 805 sequence variants and 27 494 copy number variations. We analyzed a prioritized disease-associated data set (n = 1606 variants) according to standardized guidelines, and interpreted 19 variants in 14 participants (25%) as having obvious health implications. Six of these variants (e.g., in BRCA1 or mosaic loss of an X chromosome) were pathogenic or likely pathogenic. Seven were risk factors for cancer, cardiovascular or neurobehavioural conditions. Four other variants - associated with cancer, cardiac or neurodegenerative phenotypes - remained of uncertain significance because of discrepancies among databases. We also identified a large structural chromosome aberration and a likely pathogenic mitochondrial variant. There were 172 recessive disease alleles (e.g., 5 individuals carried mutations for cystic fibrosis). Pharmacogenomics analyses revealed another 3.9 potentially relevant genotypes per individual.
Interpretation: Our analyses identified a spectrum of genetic variants with potential health impact in 25% of participants. When also considering recessive alleles and variants with potential pharmacologic relevance, all 56 participants had medically relevant findings. Although access is mostly limited to research, whole genome sequencing can provide specific and novel information with the potential of major impact for health care.
© 2018 Joule Inc. or its licensors.
Conflict of interest statement
Competing interests: Stephen Scherer serves on the Scientific Advisory Committees of Population Bio and Deep Genomics. Sherilyn Bell, Jo-Anne Herbrick, Jennifer Howe, Ann Joseph-George, Barbara Kellam, Chao Lu, Jeffrey MacDonald, Christian Marshall, Thomas Nalpathamkalam, Rohan Patel, Tara Paton, Giovanna Pellecchia, Sergio Pereira, Miriam Reuter, Stephen Scherer, Lisa Strug, Wilson Sung, Bhooma Thiruvahindrapuram, Susan Walker, Zhuozhi Wang, John Wei, Joe Whitney, Richard Wintle and Ryan Yuen have received grants from Genome Canada/Ontario Genomics; Canada Foundation for Innovation; McLaughlin Centre, University of Toronto; the Government of Ontario, Canadian Institutes of Health Research (CIHR); and the The Hospital for Sick Children Foundation during the conduct of the study. James Ellis, Matthew Hildebrandt, Hin Lee, Peter Pasceri and Wei Wei have received a grant from the McLaughlin Centre, University of Toronto. Daniele Merico is an employee of Deep Genomics. Brett Trost has received a postdoctoral fellowship from CIHR. No other competing interests were declared.
Figures
Comment in
-
Whole genome sequencing in the clinic: empowerment or too much information?CMAJ. 2018 Feb 5;190(5):E124-E125. doi: 10.1503/cmaj.180076. CMAJ. 2018. PMID: 29431109 Free PMC article. No abstract available.
-
Whole genome sequencing unlikely to be funded by government health plans.CMAJ. 2018 Apr 23;190(16):E513. doi: 10.1503/cmaj.69223. CMAJ. 2018. PMID: 29685913 Free PMC article. No abstract available.
-
The hundred-dollar genome: a health care cart before the genomic horse.CMAJ. 2018 Apr 23;190(16):E514. doi: 10.1503/cmaj.69259. CMAJ. 2018. PMID: 29685914 Free PMC article. No abstract available.
References
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous