

MEF2C Phosphorylation Is Required for Chemotherapy Resistance in Acute Myeloid Leukemia
- PMID: 29431698
- PMCID: PMC5882571
- DOI: 10.1158/2159-8290.CD-17-1271
MEF2C Phosphorylation Is Required for Chemotherapy Resistance in Acute Myeloid Leukemia
Abstract
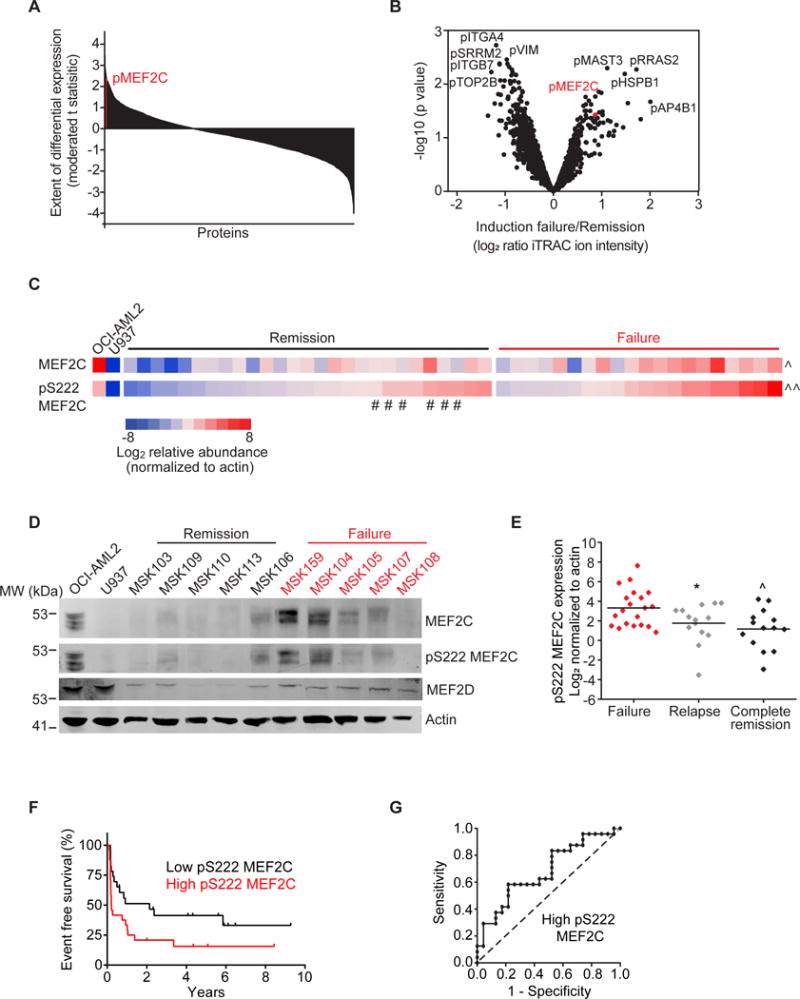
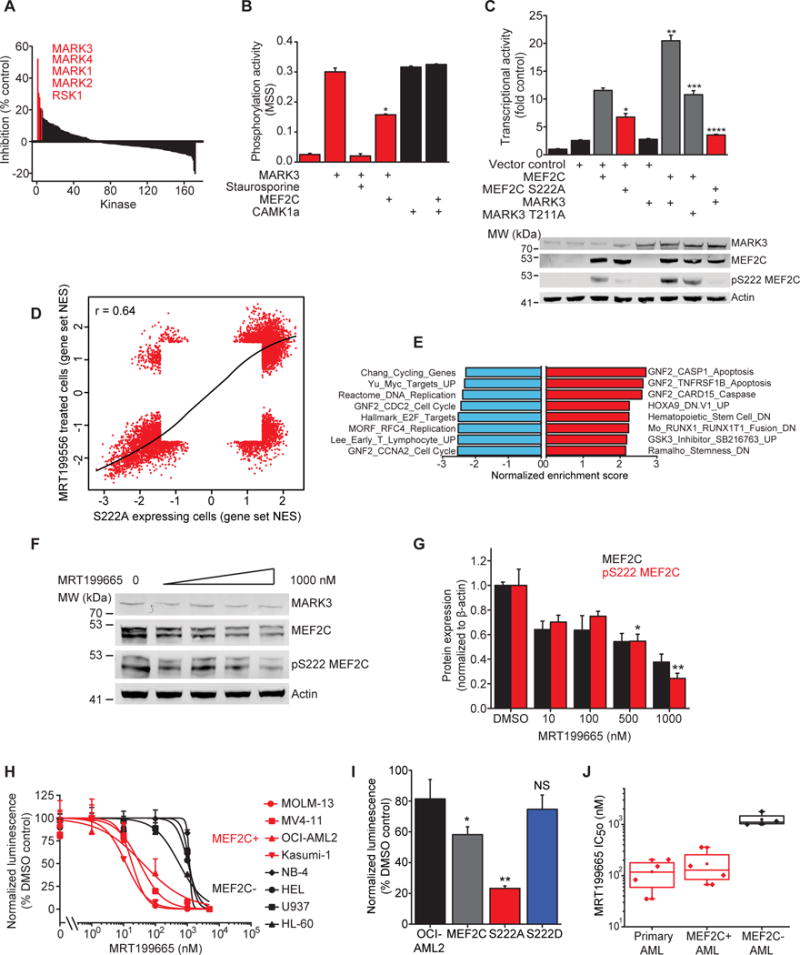
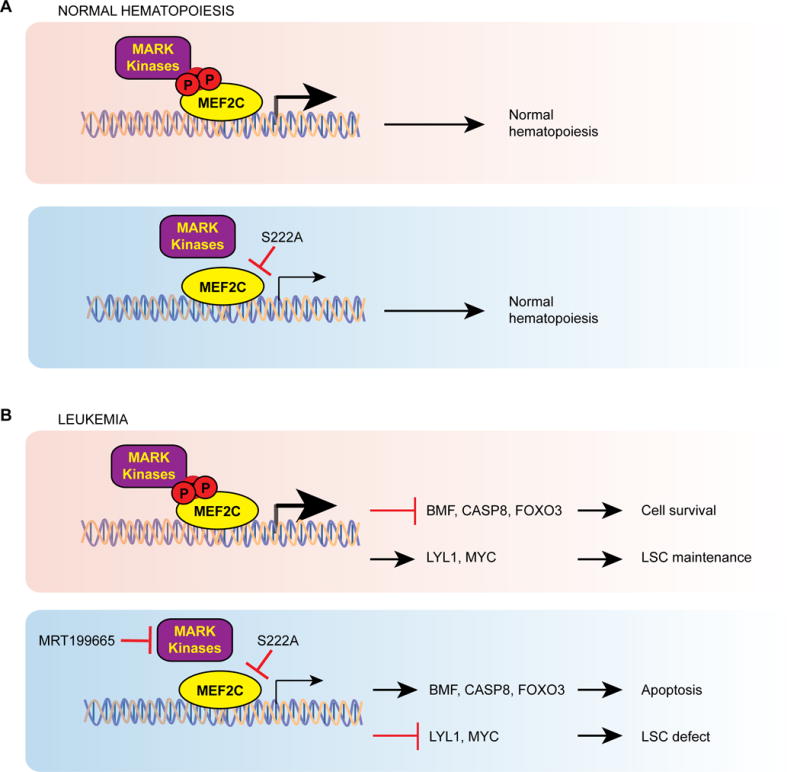
In acute myeloid leukemia (AML), chemotherapy resistance remains prevalent and poorly understood. Using functional proteomics of patient AML specimens, we identified MEF2C S222 phosphorylation as a specific marker of primary chemoresistance. We found that Mef2cS222A/S222A knock-in mutant mice engineered to block MEF2C phosphorylation exhibited normal hematopoiesis, but were resistant to leukemogenesis induced by MLL-AF9 MEF2C phosphorylation was required for leukemia stem cell maintenance and induced by MARK kinases in cells. Treatment with the selective MARK/SIK inhibitor MRT199665 caused apoptosis and conferred chemosensitivity in MEF2C-activated human AML cell lines and primary patient specimens, but not those lacking MEF2C phosphorylation. These findings identify kinase-dependent dysregulation of transcription factor control as a determinant of therapy response in AML, with immediate potential for improved diagnosis and therapy for this disease.Significance: Functional proteomics identifies phosphorylation of MEF2C in the majority of primary chemotherapy-resistant AML. Kinase-dependent dysregulation of this transcription factor confers susceptibility to MARK/SIK kinase inhibition in preclinical models, substantiating its clinical investigation for improved diagnosis and therapy of AML. Cancer Discov; 8(4); 478-97. ©2018 AACR.This article is highlighted in the In This Issue feature, p. 371.
©2018 American Association for Cancer Research.
Conflict of interest statement
No potential conflicts of interest were disclosed by the authors.
Figures




References
-
- Schuback HL, Arceci RJ, Meshinchi S. Somatic characterization of pediatric acute myeloid leukemia using next-generation sequencing. Seminars in hematology. 2013;50:325–32. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
